-
输入文件格式
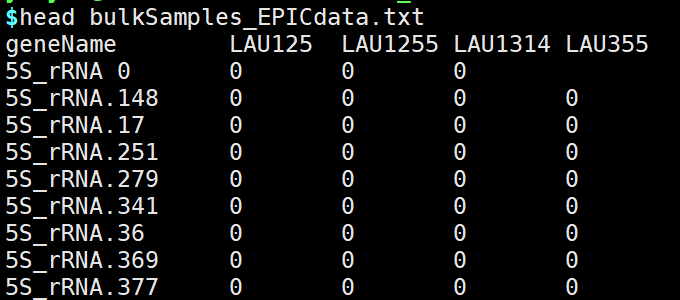
-
处理之后的输入格式
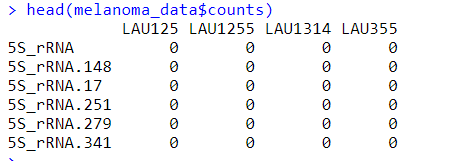
-
使用脚本
args=commandArgs(T)
input <- args[1]
output <- args[2]
#1.默认参数
library(EPIC)
#读入表达量矩阵
df = read.table(input,sep=" ",header = T)
#设置行名
row.names(df)=df[,"geneName"]
#去掉基因名所在列
df <- subset(df,select = -c(geneName))
#使用默认的细胞类群
res1 <- EPIC(df)
#输出细胞在不同样品的比例的结果
write.table(res1$cellFractions,output,sep=" ",col.names = T,row.names = T,quote = F)
- 使用命令
Rscript EPIC.R bulkSamples_EPICdata.txt cellFractions2.txt
-
输出文件结果
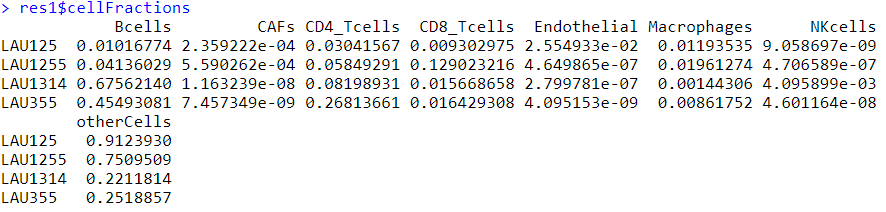
-
输出文件格式
