# Thanks .这是scut系统发育学课程论文。
1、简介:
进化生物学是指研究不同生物在一段时期中的改变,其目的是提供理论,进而解释生物多样性的过程和机制。与证据充足的生物学相比,支持进化生物学的确凿证据并不多,往往只能通过其他生物学分支学科提供的例证来形成理论,形成的理论来解释这些例证。譬如鲸类为什么能适应水生环境,通过生物学上基本特征的观察:毛发、骨盆的消失和减少,进而产生鲸类的祖先可能直接起源于毛发少的两栖动物而非地球上的哺乳类的假设,这些假设并不直观,往往十分错综。
涉及进化的分子生物学问题也是复杂的:基因随机突变产生基因的多态性,但是这种基因的多态性是如何在种群中得以有效的保存呢?通常的解释是自然选择(压力),对突变的正负选择,使得突变得以保存或淘汰;另一种解释是中性突变占绝大多数,多态基因是否保存取决于新基因的产生与通过遗传漂变而使得基因消失之间的平衡。
进化生物学产生了很多分支,如分子进化主要是研究DNA、RNA、氨基酸等生物大分子的进化历史,系统发育学主要研究有机体的多样性以及它们的关系和类别。(我们大可不必纠结与具体的概念,只需有进化的思想。如题,序列比对提示我们用的往往是DNA等生物大分子序列,故分子进化=分子系统发育。)
进化生物学的应用:农业上可以通过人工选择,提高作物的遗传多样性;医学上可以发现病原生物与其寄主共同进化的关系,大剂量的抗生素的滥用,有可能使得耐药性细菌得以选择,进而使得药物失效。很多生物学过程与机制都可以用进化的眼光去解释(如,CRISPR/Cas9中的噬菌体与细菌对抗的选择机制)。
2、进化理论
2.1 进化历史:
自然选择理论:拉马克、达尔文,通过化石、同功器官或者其他资料佐证。
2.2 分子进化理论:
两个基因之间的差异称为趋异度:可用单位进化时期来表示,即产生1%趋异度所用的时间。
 分子钟是指:蛋白质氨基酸之间的趋异度与其分开后的时间成正比,每一种蛋白质的相对恒定进化速率。
分子钟是指:蛋白质氨基酸之间的趋异度与其分开后的时间成正比,每一种蛋白质的相对恒定进化速率。
中性进化理论 :
生物进化基于:遗传变异的产生和后代中的变异分选
DNA序列中的变异来自随机突变:同义突变 错义突变(无义突变)
中性突变:对表型没有影响的突变为中性突变:遗传漂变是指中性突变变异体频率的随机变化过程
非中性突变:容易受到自然选择:负选择消除新突变,当变异体逐渐去除,群体内几乎没有变异;正选择为有利表型,当新突变最终取代原有序列时,自然选择降低了群体内的变异,但是可能在两个群体之间产生更大的变异。
同义和非同义氨基酸替换的比值P可以衡量正负选择:P>1正选择:免疫球蛋白的肽结合区 P=1 中性进化 P<1 负选择
中性进化学说认为:大多数突变基因为中性突变,中立突变基因在群体的固定是随机漂移引起的,因此功能上重要的基因受到更多的选择压力而进化速率慢,新基因的产生主要以基因重复和不等交换方式进行。
2.3 分子进化与传统进化的理论区别与联系:

(注:图片仅供学习,侵权删!)
3、分子系统发育分析(phylogeny analysis)
3.1 概念和分析步骤
<1> 系统发育学研究的是进化关系,推断或者评估单位之间的进化关系。通过系统发育分析所推断出来的进化关系一般用进化树来描述,这个进化树就描述了同一谱系的进化关系,包括了分子进化(基因树)、物种进化以及分子进化和物种进化的综合。
<2>为什么使用系统进化树?

鉴定未知的物种亲属关系最近的类型:如用核糖体RNA;通过进化树直系同源关系,发现新的功能基因(不同物种);重现基因起源,揭示一些基因晚期的变化。(直系同源orthologous与旁系同源paralogous:汽车轮子与轮子直接是旁系同源,轮子与转盘是直系同源。)
<3>主要步骤有呢?
数据收集,多序列比对,数学建模,进化树构建,检验评估

3.2 数据选取
下面主要讲下数据选取与其对应构建的树:

(基因水平转移)同源基因建的树homologous genes称为基因树(gene tree);paralogous (旁系同源),基因家族树;orthologous (直系同源)——不同species,species tree,eg:核糖体RNA广泛存在于各类物种,可用它构建生命之树。
3.3 序列比对:
(一) 原理概述:
联配,比较相似性,进行同源性分析(反映共同祖先序列进化):可用于搜索相似序列,预测。
大概提下Needle-Wunsch算法全局比对算法、Smith-Waterman局部比对算法:引入计分方法(匹配成功+1,错配-1,匹配空格-2,空格之间匹配0);每次使局部分级最优,最后累达到总的最优化。
(二)多序列比对:1)、累进法:序列两两比对,产生向导树,加入序列依次比对(进化距离为负分总和绝对值)2)、星比对法:两两比对后,选中心序列,将其他序列与中心序列对齐。
(三)比对结果:使用ClustalW( www.ebi.ac.uk/clustalw)进行多序列比对
最后对比对序列进行改善,如下;
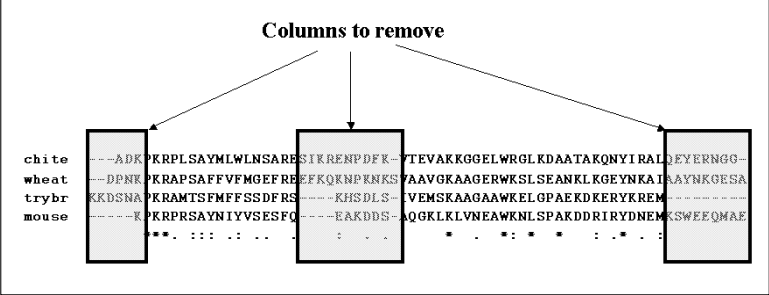
去除不C、N端、不匹配的区域(gap)较多的序列处也去除。
3.4 数学模型选择:
DNA序列进化就是位点上的核苷酸随时间的变化,包括:碱基替换、缺失和插入。根据不同进化改变类型构建的模型有替换模型(substitution model)和indel 模型。
蛋白质序列数学模型考虑氨基酸取代速率、组成、速率变异,有经验模型、机理模型、固定速率模型与可变速率模型:广义时间可逆取代模型(general time-reversible model)允许氨基酸组成频率和取代速率自由变化。
3.5 进化树:
3.5.1 进化树
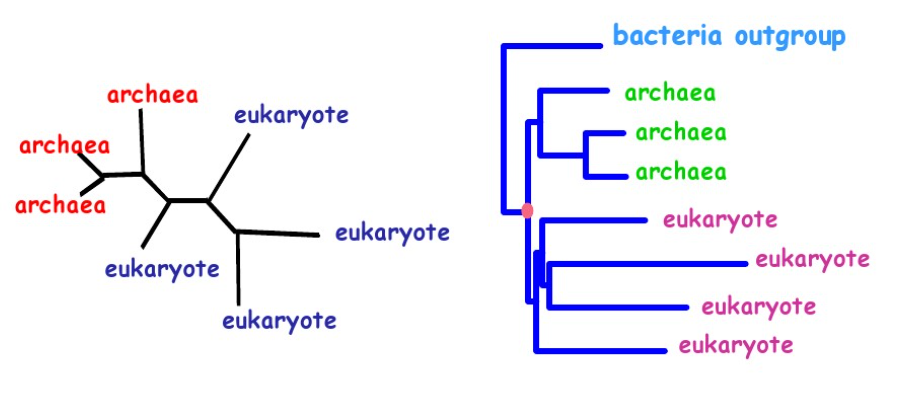
3.5.2 进化树构建方法
1、距离法:UPGMA(非加权组平均法):基于遗传距离聚类
邻接法(NJ):选星状树,算距离,合并;
选邻居,算距离,合并;
... ...
2、最大简约法(MP):根据信息位点(有序列元素比对变化)提供的各序列替换情况,在所有可能的树中寻找含有最小替换数的树的方法。(蛋白质的PAM矩阵时,计算替换数也会用到这种方法)
3、最大似然法(ML):所有可能的树中所以可能的替换数方式中,选择可能性最大的一种方式(引入了一个基于先验的似然函数,最后求得似然率最大时的枝长)。
4、贝叶斯法:后验概率法
3.6 进化树评估
自举检验(Bootstrap Method):就是一种抽样检验
首先从排列的多序列中随机有放回的抽取某一列,构成相同长度的新的排列序列;重复,得到多组序列;最后对这些新的序列进行建树,观察这些树与原始树是否有差异,以此评价建树的可靠性。
模拟研究表明,在合适的条件下,亦即各种替换速率基本相等,树枝基本对称的条件下,如果自举值大于70,那么系统发育进化树能够反映真实的系统发生史的可能性要大于95%。
4、系统发育分析软件
4.1 序列准备与多序列比对
4.1.1 DNA(https://zhuanlan.zhihu.com/p/36598434 知乎白石墨)
(1)下载fasta格式序列
输入你想查找的序列,比如Syp基因:

(2)进入基因详细页面:
(3)点击左下角GenBank,进入点击send to:下载fasta格式序列
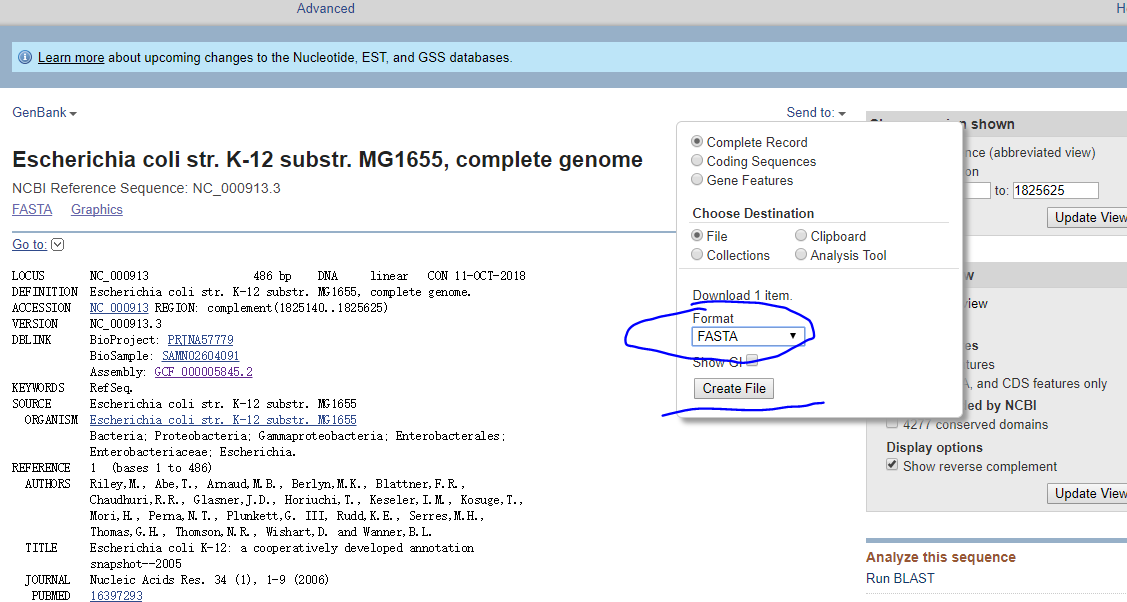
于是我选了12条序列(1 2 3 5 6 7 8 9 10 11 12 14):下面合并这些序列于一个fasta文件中:

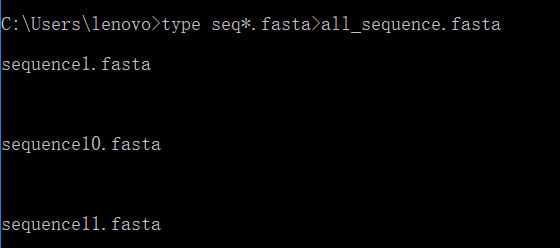
得到整合文件 all_sequence.fasta.
4.1.2 Protien
NCBI查找 gamma fibrinogen(纤维蛋白).氨基酸序列 (UniProtKB/Swiss-Prot)
O12957 (Sheep), O02672 (Moose), O02683 (Giraffe)
O02690 (Chevrotain), O02681 (Beluga) O02687 (Sperm_whale)
O02673 (Rorqual), O02688 (Pig), O12959 (Peccary)
O02677 (Dromedary), O02689 (Tapir), O02682 (Horse)
O02676 (Hyena), O02680 (Coyote), O12954 (Hippopotamus)
>sheep RFGSYCPTTCGIADFLSNYQTSVDKDLRNLEGIFYQVENKTSEATELVKAIKISYNPDEPSKPSNIESATKNYKRMM
>Moose
RFGSYCPTTCGVADFLSNYQTSVDKDLQNLEGILYQVENKTSEARELVKAIQISYNPDEPSKPNNIESATKNSKRMM
>Giraffe
RFGSYCPTTCGVADFLSNYQTSVEXDLYRLESDLYQVENKTSEAKELVKAIQISYNPDEPSKPSHIESATKNSKRMM
>Chevrotain
RFGSYCPTTCGIADFLSNYQTSVDKDLHNLESILYQVENKTSEARELVKAIQISYNPDEASKPNKIESATRNSKKMM
>Beluga
RFGSYCPTTCGIADFLSXYQTSVDKDLQNLEGILRQVENKTSEARELVKAIQISYRSDGPAKPNGIESATKISKKVL
>Sperm_whale
RFGSYCPTTCGIADFLSTYQTNVDKDLQNLEGILRQVENKTSEARELVKEIQISYRSDGPAKPSGIESATKNSKKML
>Rorqual
RFGSYCPTTCGIADFLSTYQTSVDKDLQNLEGILRQVENKTSEARELVKAIQISYRSDGPAXPNGIDSATKISKKML
>Pig
RFGSYCPTMCGIAGFLSTYQNTVEKDLQNLEGILHQVENKTSEARELIKAIQISYNPEDLSKPDRIQSATKESKKML
>Peccary
RFGSYCPTTCGITDFLSTYQNTVEKDLQNLEGILHQVENKTSEAKELIKAIQISYNPDXPSKPDRIQSATKDSKKML
>Dromedary
RFGSYCPTTCGIADFLSTYQNSVDKDLQTLEDILHQVENKTTEARELIKAVQISYNPAEPSKPSRIESATKDFKKMM
>Tapir
RFGSYCPTTCGIADFLSTYQTXVDKDLQVLEDILNQAENKTSEAKELIKAIQVRYKPDEPTKPGGIDSATRESKKML
>Horse
RFGSYCPTTCGIADFLSNYQTSVDKDLQDFEDILHRAENQTSEAEQLIQAIRTSYNPDEPPKTGRIDAATRESKKMM
>Hyena
RFGSYCPTTCGIADFLSTYQTGVXNDLRTLEDLLSGIENKTSEAKELIKSIQVSYNPNEPPKPNTIVSATKDSKKMM
>Coyote
RFGSYCPTTCGIADFLXTYQTGVDNDLQALEDLLRRIENKTAEAKEVIKSIQITYNPDEPPKPNRVVGATXDSKKMM
>Hippopotamus
RFGSYCPXTCGVADFLSNYXTSVDKDLQNLESIVHEVENKTSEARELVKAIQISYNPDEPEKPSRIESATKNSKKMX
4.1.3 多序列比对
使用ClustalX 2.1 进行多序列比对:
File-load 载入合并后的文件all_sequence.fasta.

点击Alignment-Alignment Parameters :一般默认就行(23333)
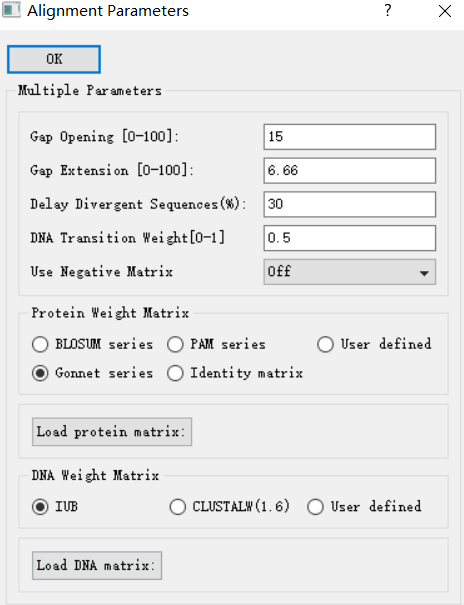
点击Output Format option :勾选PHYLIP 格式 点击ok

点击 Do Complete Alignment ,程序运行,输出 aln、dnd(前导树文件,和进化树很像)、phy后缀的三个文件。
结果需要编辑第一列只留下物种名,序列去掉5',3'端的空序列(因为要比对序列同源性,最好把显示 - 的序列去掉,使多序列的两端整齐,类似矩阵)
4.2 MEGA
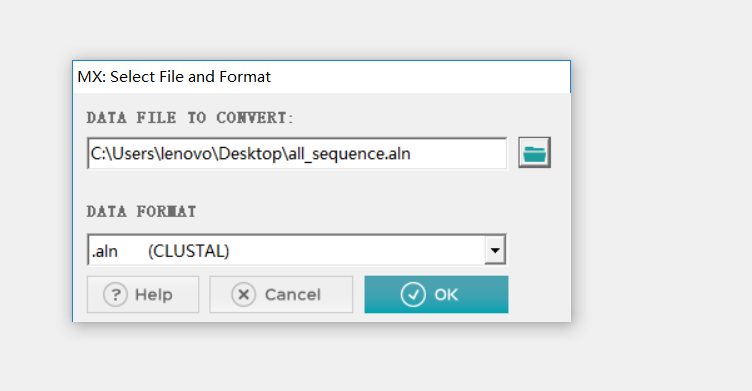
(1)打开MEGA-X,将aln格式文件转换成Mega格式:得到all_seq.meg格式文件
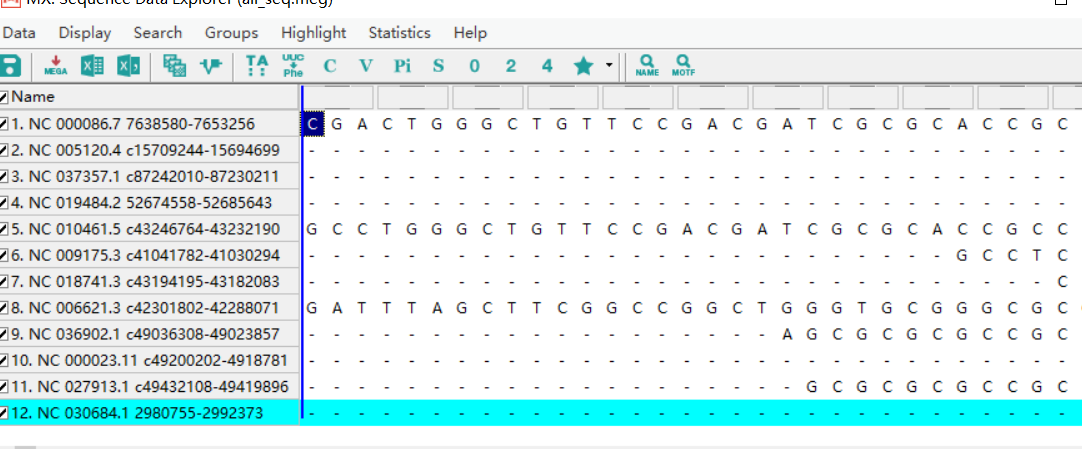
(2)导入meg文件:选择Nucleotide sequences。

(3)PHYLOGENY:选择建树方法(构建ML树)
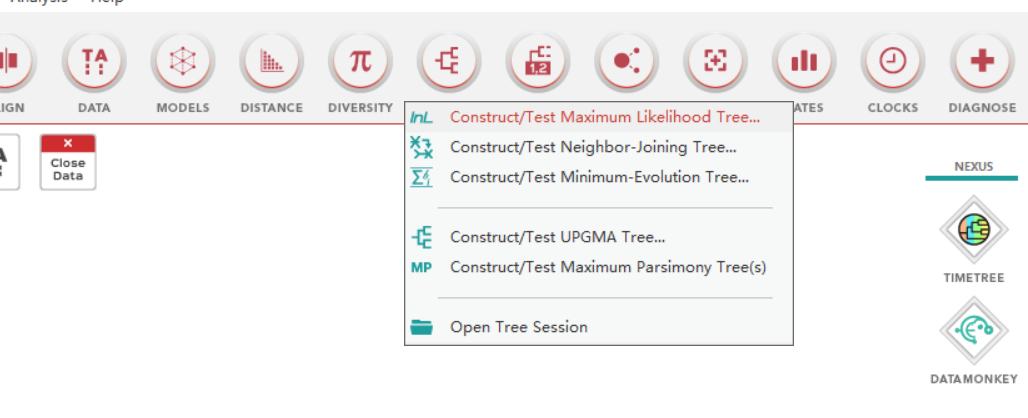
建树方法、分子数检测方法、碱基替换数学模型。若序列为protien可以选取:广义时间可逆取代模型(general time-reversible model)。
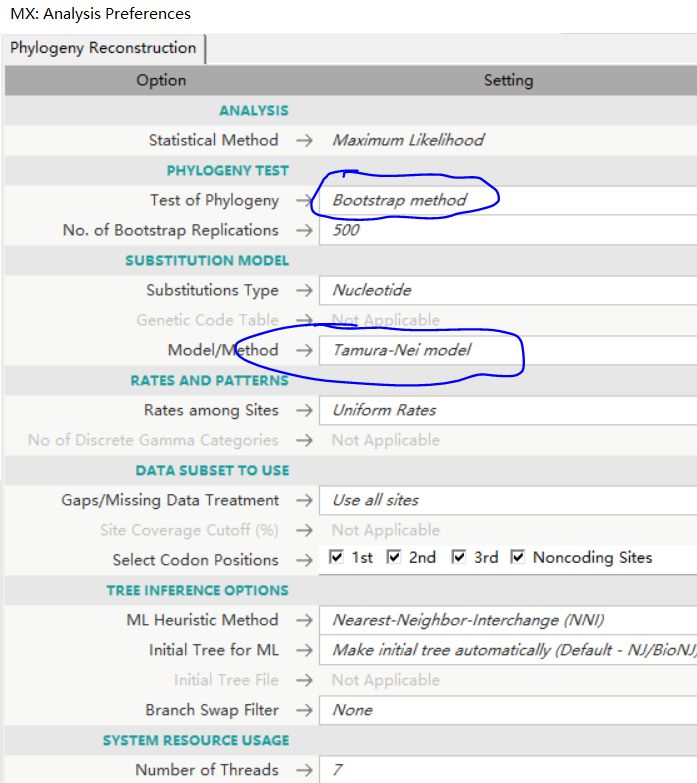
程序运行过程:

获得进化树:窗口有两个属性页,一个是Original tree ,一个是bootstrap 验证过的一致树,bootstrap值大于70的话,一般情况下有95的可能性重构进化过程。
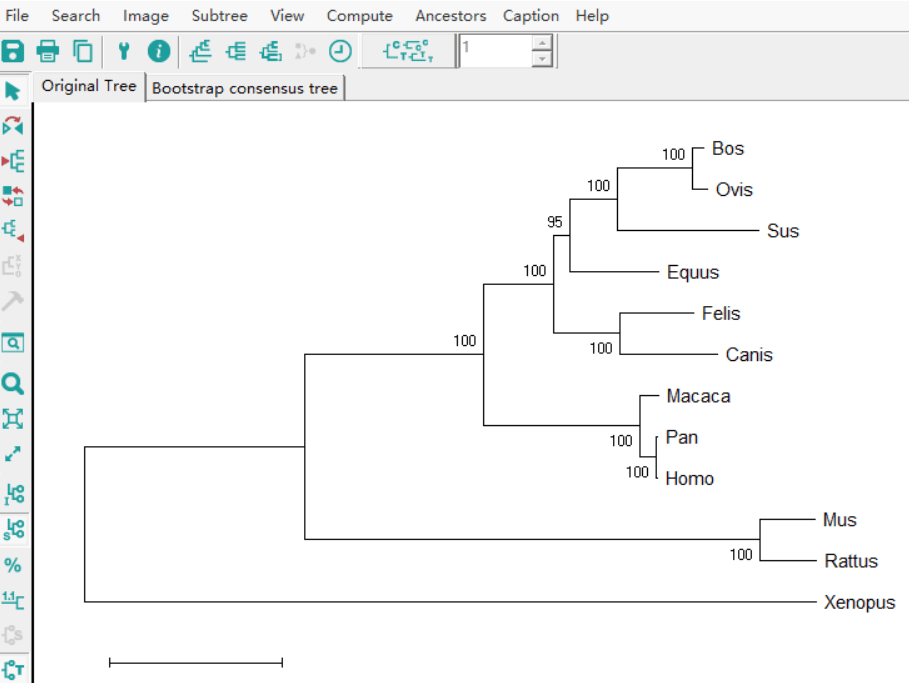
可以使用多种建树方法可得出不同的树,根据自举值对进化树进行优化,这里不讨论进化树的生物学意义。
4.3 Phylip
Phylip是一个免费的系统发育构建软件包:按照建树所需使用软件包来infile-outfile的过程
软件包如下:常用的有seqboot dnadist protdist consense


(1)PHY文件:ClustalX 2.1导出的*.phy文件格式有sequence和interleaved两种。这次使gamma fibrinogen氨基酸序列来建树。文件第一行包括两个数:第一个数为欲分析的序列数第二个为各组分析的碱基或氨基酸数(最少的)。
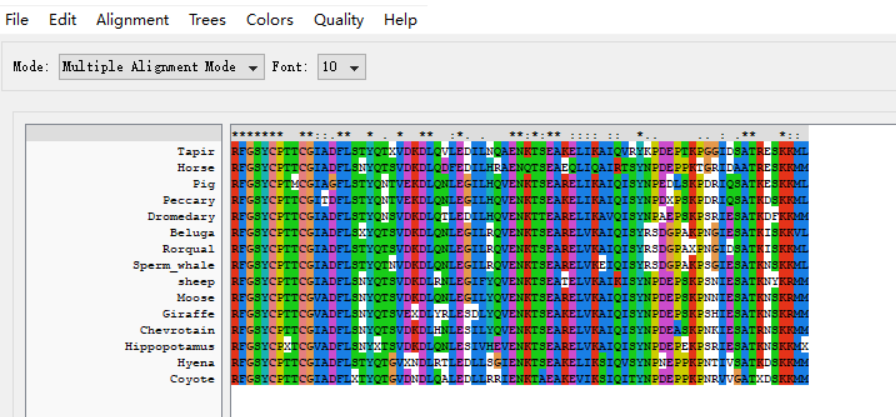
clusterX比对结果显示蛋白同源性
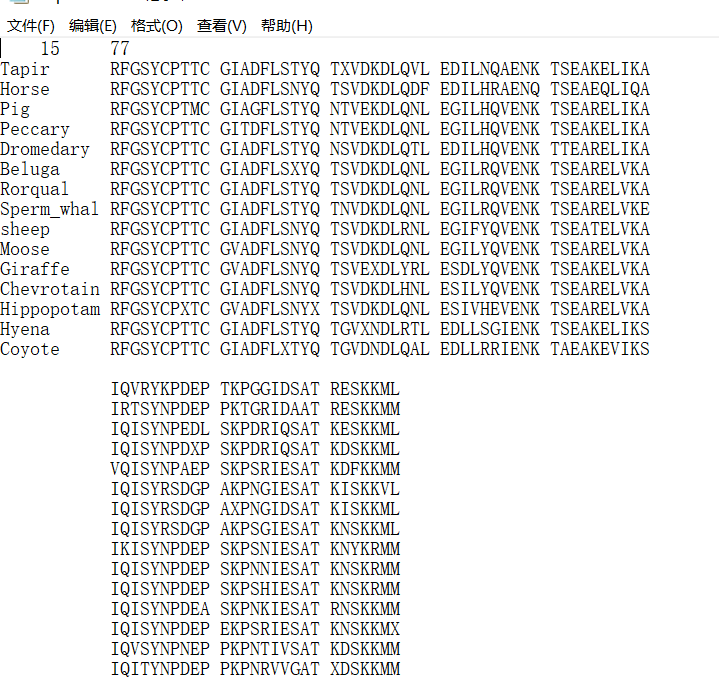
interleaved文件格式
(2)运行seqboot程序:
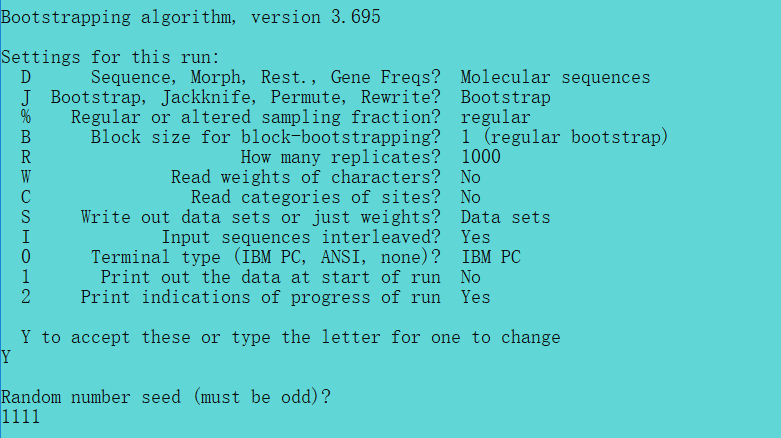
1)首先将获得的*.phy数据名改为:seqbootinfile.phy,输入数据文件名:C:GFseqbootinfile.py

2)程序需要输入一个随机数(4n+1),输入1111,选项包括数据套大小(100-1000),检测方法;程序运行之后得到一个outfile的文件;最后enter键退出。
(3)运行Prodist:距离法,将上一步的outfile改成prodistinfile;打开Prodist程序(使用NJ法构建进化树,如使用其他算法,则使用其他软件),输入m,回车;输入D,回车,输入1000(其实默认就行);输入Y,最后获得一个outfile。

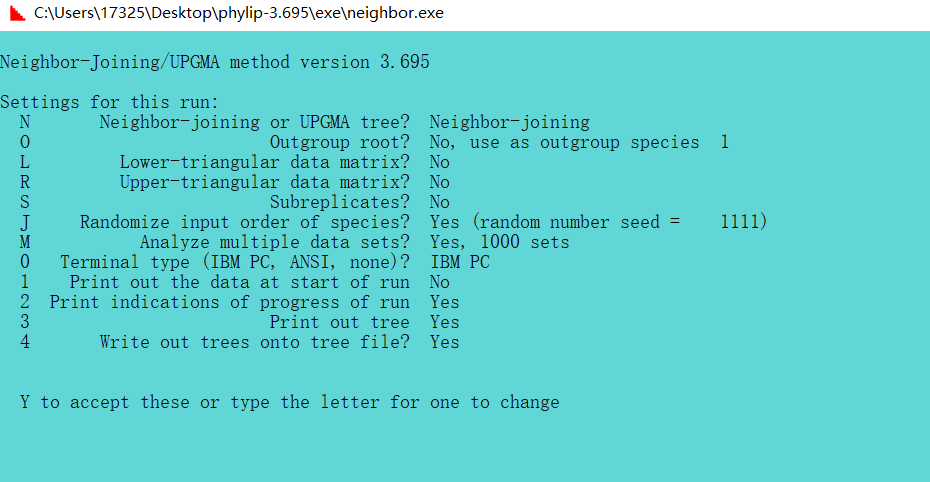
(4)运行neighbor程序:使用NJ法建树,将上步outfile改为neighborinfile;打开程序,键入文件,m,回车;键入1000;奇数1111,回车;Y,运行一段时间得到outfile和outtree:outfile是分析结果的输出报告,outtree可用treeview打开。
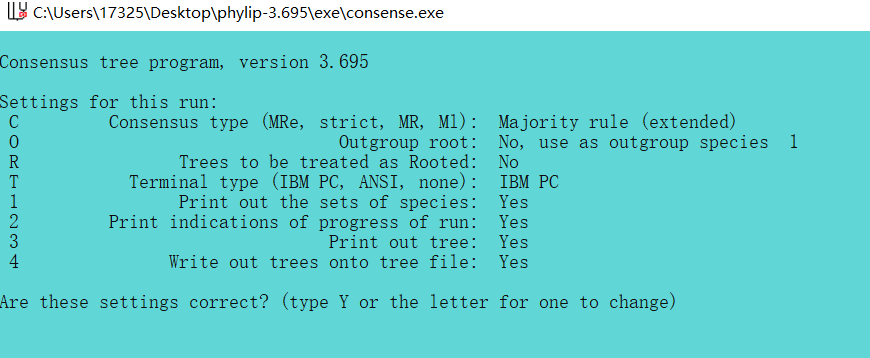
(5)运行consense程序:构建一致树,将outtree文件命名为consenseintree:打开程序键入文件名,最后得到的outtree就是最终结果,可用treeview打开,我这儿用的是figtree这个软件打开的。
(6)最终的outtree:使用figtree打开,figtree软件可对其微调,这里没有进一步去美化以及探讨生物学意义了:该进化树反映了sigame纤维蛋白的进化关系。

(7)总结:Phylip构建发育树就是一个filein-fileout的过程;根据不同的序列选择不同的程序,来建模与构建。
 系统发育树分析用到的文件
系统发育树分析用到的文件
4.3 进化树的美化
进化树美化以及整合其他数据集——后期以及进一步展示所要做的事情,这儿贴几个美化的图来结束吧。
进化树与箱线图:
 进化树与热图:
进化树与热图:
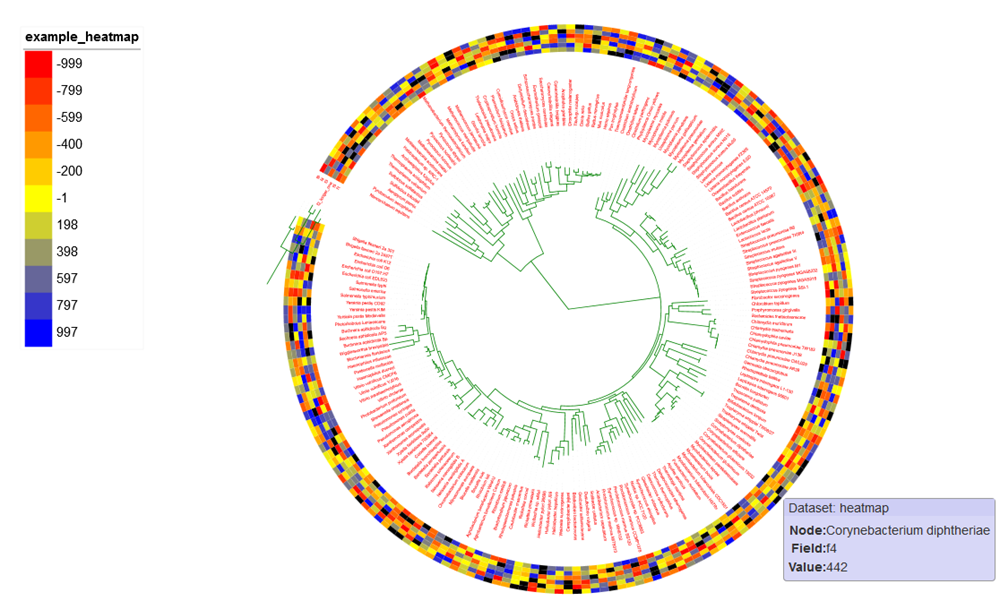
链接如下:
iToL (https://www.plob.org/article/12745.html)
ggtree(https://www.plob.org/article/12853.html)
5、最后:感谢网上文档的提供者,以及论坛的资料,参考太多东西了,很乱,版权侵犯请联系我删帖:本资料仅供个人学习交流。
