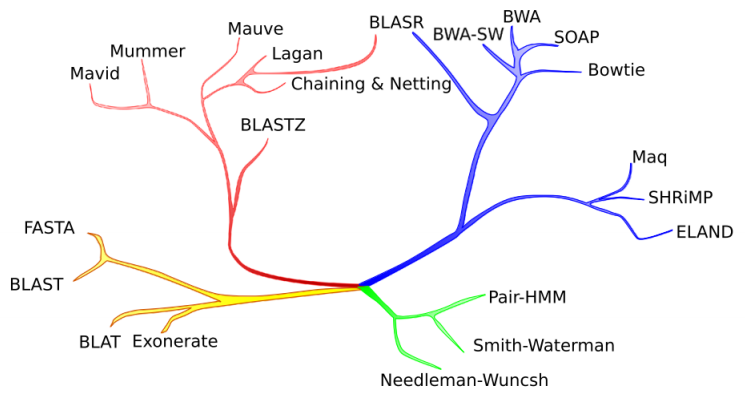
(green) short pairwise alignment / detailed edit model;
(yellow) database search / divergent homology detection;
(red) whole genome alignment / alignment of long sequences with structural rearrangements;
(blue) short read mapping / rapid alignment of massive numbers of short sequences.
简单的局部比对:Blast、Blat等
Blast类型包括以下几种:
Blastn---核酸序列与数据库中的核酸序列的比对
Blastp---蛋白序列与数据库中的蛋白序列的比对
Blastx---核酸序列翻译成蛋白质(six-frame protein)后与数据库中的蛋白序列进行比对
Tblastn---蛋白质序列与数据库中的核酸序列进行比对
Tblastx---核酸序列翻译成蛋白质后与数据库中的核酸序列翻译成的蛋白质序列进行比对
BLAST建库
建库的过程是建立目标序列的索引文件,所用程序是formatdb,通常我们使用FASTA格式的序列作为输入。用于建库的FASTA序列是db.seq,formatdb的基本命令是:
formatdb -i db.seq [-options]
常用参数有以下几个:
-p:选择建库的类型,T表示蛋白库,F表示核酸库。缺省值为T
-o:判断是否分析序列名并建立序列名索引。T表示建立序列名索引,F表示不建立序列名索引。缺省值为F
BLAST命令行:

输出结果:
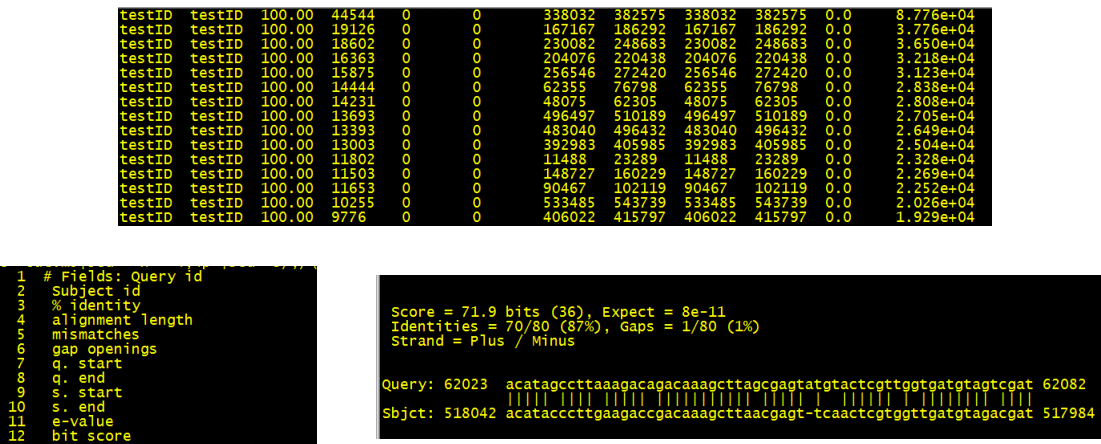
3是相似度、4是能比对上的区域
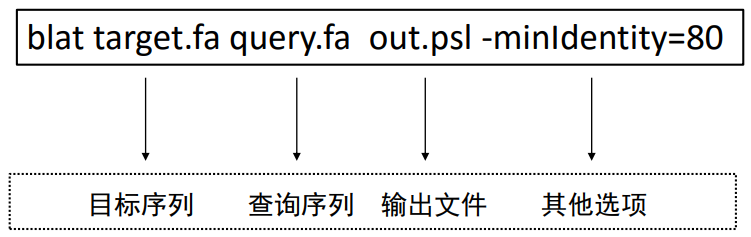
Blat
全局比对:MUSCLE、MATT等
muscle -in <inputfile> -out <outputfile>