一句话评价:小样本量单细胞转录组在冷门疾病领域应用前景尚可,常规套路也能发NC
1. Title

标题可以看出本研究的主要内容:①构建成纤维细胞的图谱/揭示成纤维细胞的异质性;②找到一群(相对于正常样本)在纤维化皮肤病中含量升高的间充质成纤维细胞
2. Background

- 纤维化的特点是成纤维细胞增殖、迁移、侵袭能力增加以及胞外基质 (ECM) 的过度积累,在全球范围内都有较高的发病率和死亡率。 纤维化皮肤病主要是 ECM 在真皮中积累
- 纤维化可影响任何器官,导致进行性组织瘢痕形成和器官功能障碍
- 迄今为止,纤维化皮肤病的发病机制尚未完全阐明,仍缺乏根治性治疗方法
- 尚缺乏关于 scRNA-seq 探索纤维化皮肤病成纤维细胞异质性的研究
——> 所以要做single cell
3. Results
3.1. 单细胞测序揭示正常伤疤和纤维皮肤病瘢痕的(真皮层)细胞异质性
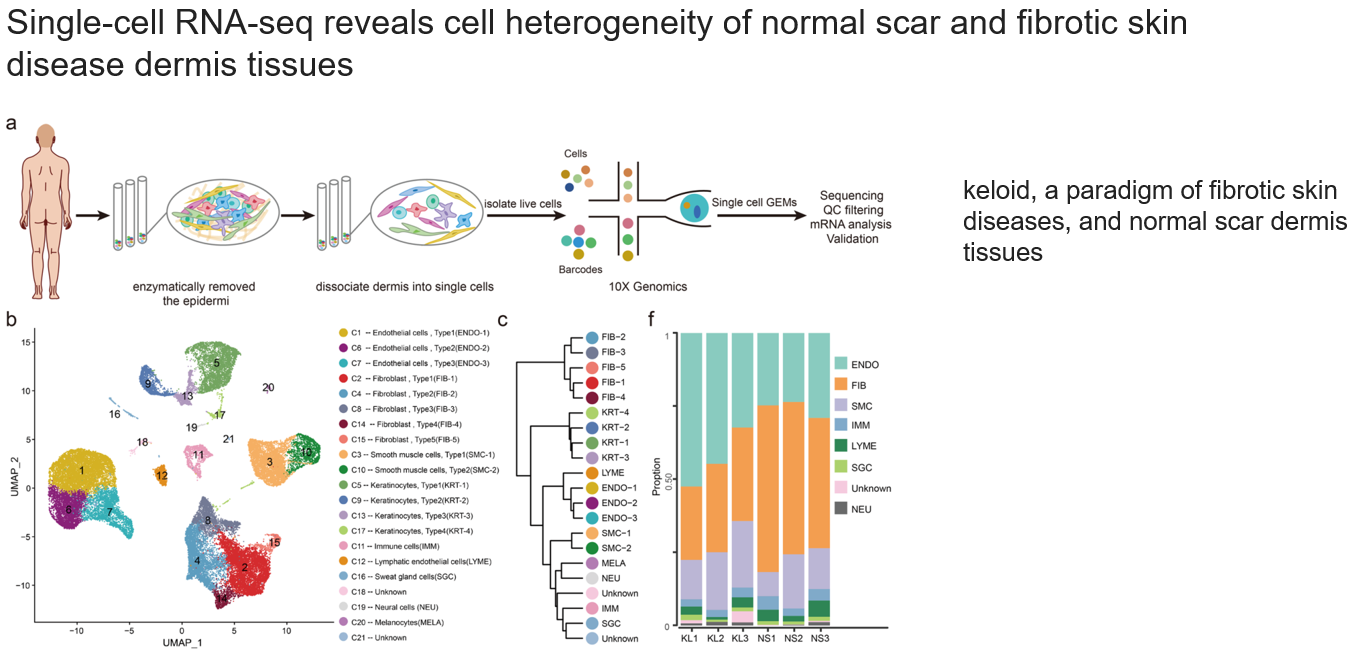
keloid在此处表示瘢痕/“病灶”
都是很常见的图:
- a.流程图,比一般情况多了酶解这一步,目的是消除表皮层的细胞
- b.umap降维,展示cluster注释信息
- c.层次聚类,表示cluster表达谱的相似性
- d.各种细胞组分在每个样本中的占比
3.2 成纤维细胞被分为4群,其中间充质类型的成纤维细胞在疾病组织中增多
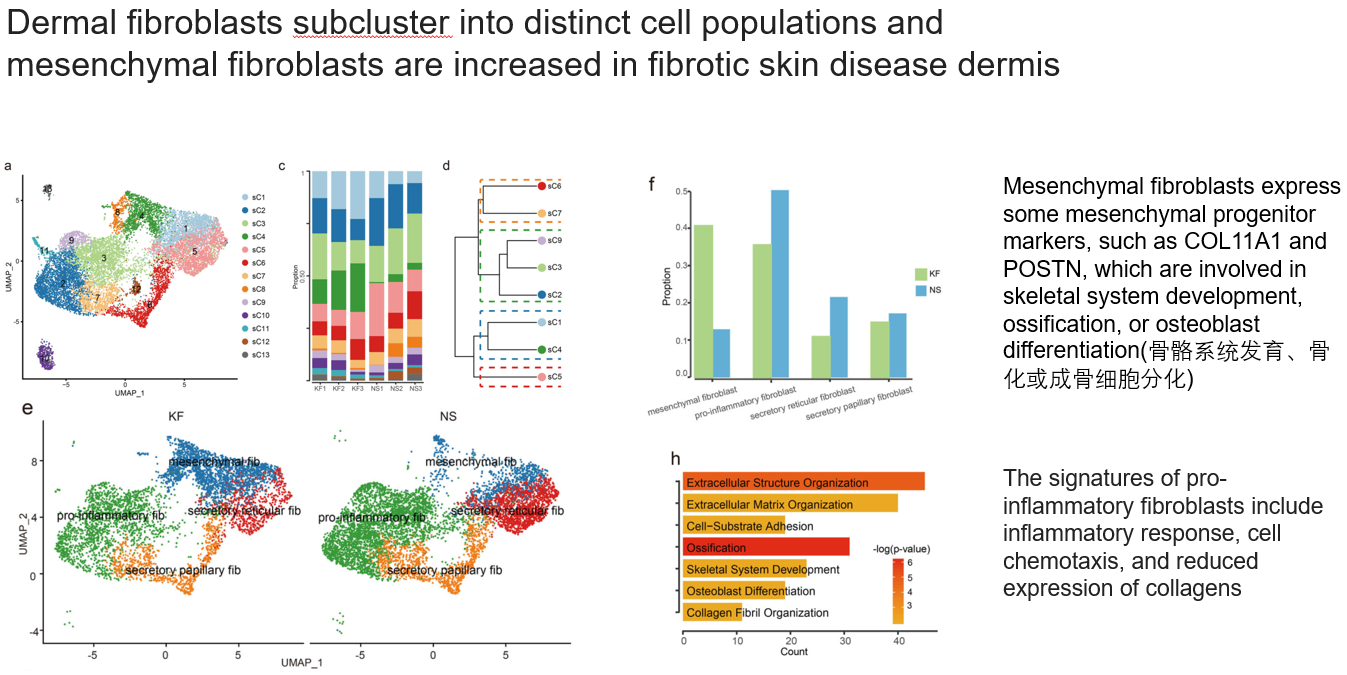
- a, c, d, e. 在上一步的基础上,提取出成纤维细胞重新降维, 聚类。结合已有文献,最终注释为4群,其中间充质成纤维细胞表达一些间充质祖细胞marker,如 COL11A1 和 POSTN,它们参与骨骼系统发育、骨化或成骨细胞分化。促炎成纤维细胞的特征包括炎症反应、细胞趋化性和胶原蛋白表达减少
- f. 只有间充质类型的成纤维细胞(Mesenchymal fibroblasts,MF)在疾病组织中升高
- h. 对比这种MF在疾病、正常之间的差异,发现疾病状态下,MF高表达骨骼系统发育、骨化相关通路和基因,比如COL11A1, COMP, POSTN
不知道大家读到这里有没有跟我一样的困惑:①某个细胞类型和其他细胞类型做对比,②这种细胞内部不同condition做对比,得到的差异基因和通路这么像?!
3.3 疾病组织中,间充质成纤维细胞的特征
(也就是在疾病组织中,将MF和其他成纤维细胞做对比)
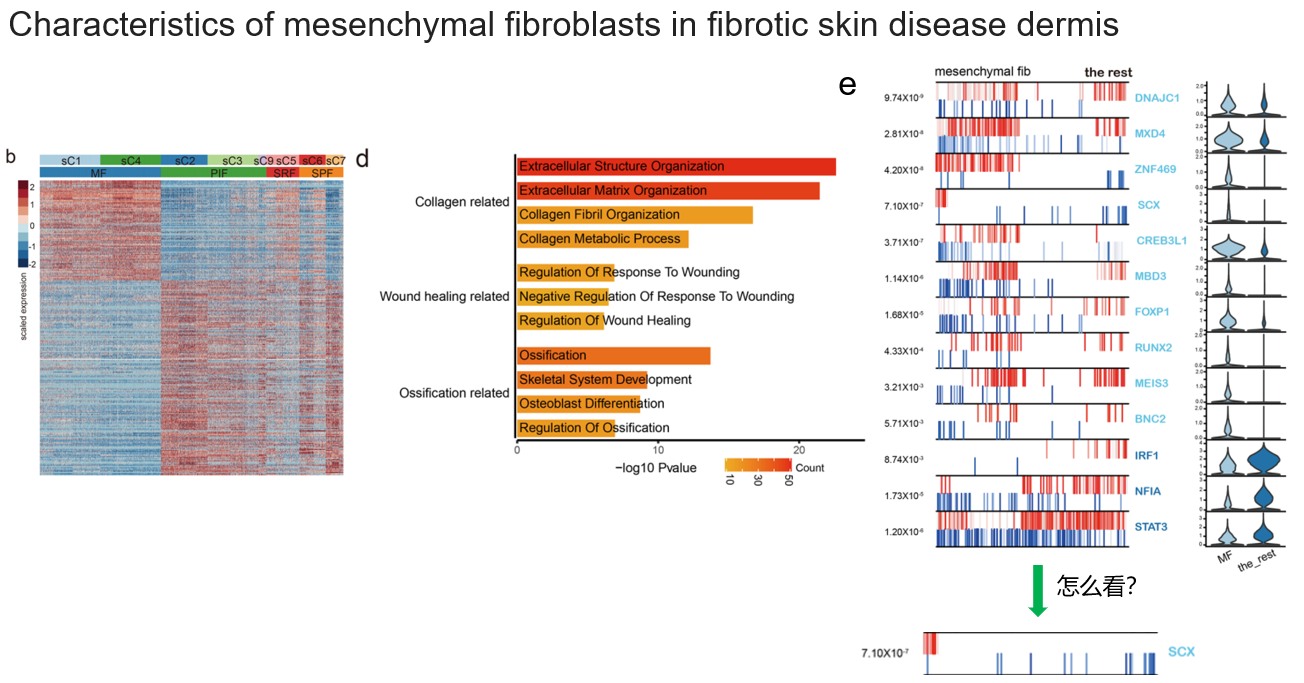
- b. 差异基因
- d. 差异通路主要分了三块:胶原蛋白(ECM的成分)相关、伤口愈合相关、骨化相关
- e. 这个图是看的转录调控,并没有采用常见的单细胞转录组中分析转录调控的方法。右边的小提琴表示TF在MF与the rest之间的表达差异。左边的图,每个TF对应一行,一行里面有两小行,列表示按照logFC排序后的基因,越往左表示MF中高表达的基因,越往右,表示the rest中高表达的基因。如果某个调控网络(TF加靶基因)在某个群中存在,则应该满足:TF高表达、TF正向调控的基因高表达、TF负向调控的基因低表达。以SCX为例,它在MF中高表达,它激活的基因(红色表示)在MF中高表达(因为这些基因排在左边),它抑制的基因(蓝色表示)在MF中低表达(因为这些基因排在右边),结合这几点共表达信息,就支持了MF中存在SCX的调控网络。通过这个分析,找到了MF中特有的调控网络,并和它上调的通路是吻合的。
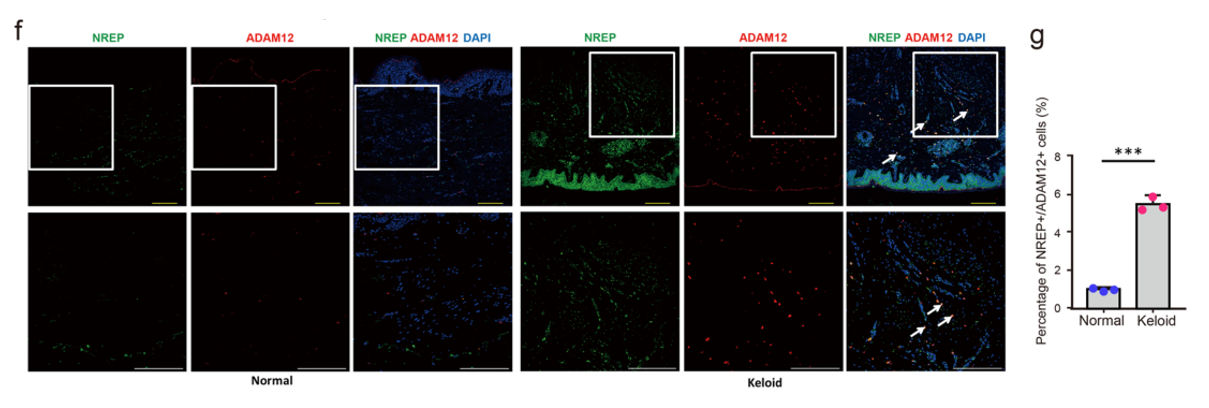
- 荧光染色验证这群MF在疾病组织中存在,在正常组织中很少
3.4 细胞通讯分析

- 找到了疾病组织中显著的interaction
- 探究MF在细胞互作中所起的角色,MF有哪些重要的interaction
3.5 疾病真皮组织中MF通过POSTN促进胶原蛋白的表达

接着是做了一点点机制。关于为什么要做这个基因,正文没有说太多,只说是:①这个基因在MF中高表达;②此前已有一些报道称POSTN可以促进瘢痕组织的形成,可以促进胶原蛋白(一种ECM成分)的产生。
- a. 通过单细胞转录组分析中得到的marker,将成纤维细胞分选为MF和other fibroblasts
- d. 对这两组细胞做了bulk RNA-seq,证明分选效果比较好,得到了想要的细胞

- g. 将两组细胞培养后,取上清液加到other fibroblasts里面,发现MF的上清液能使两种胶原蛋白表达升高
- h. 加入POSTN的中和抗体后,这种现象被抑制了
通过一正一反的实验就证明了MF通过POSTN促进胶原蛋白表达的结论
3.6 MF的两个子类有什么关系
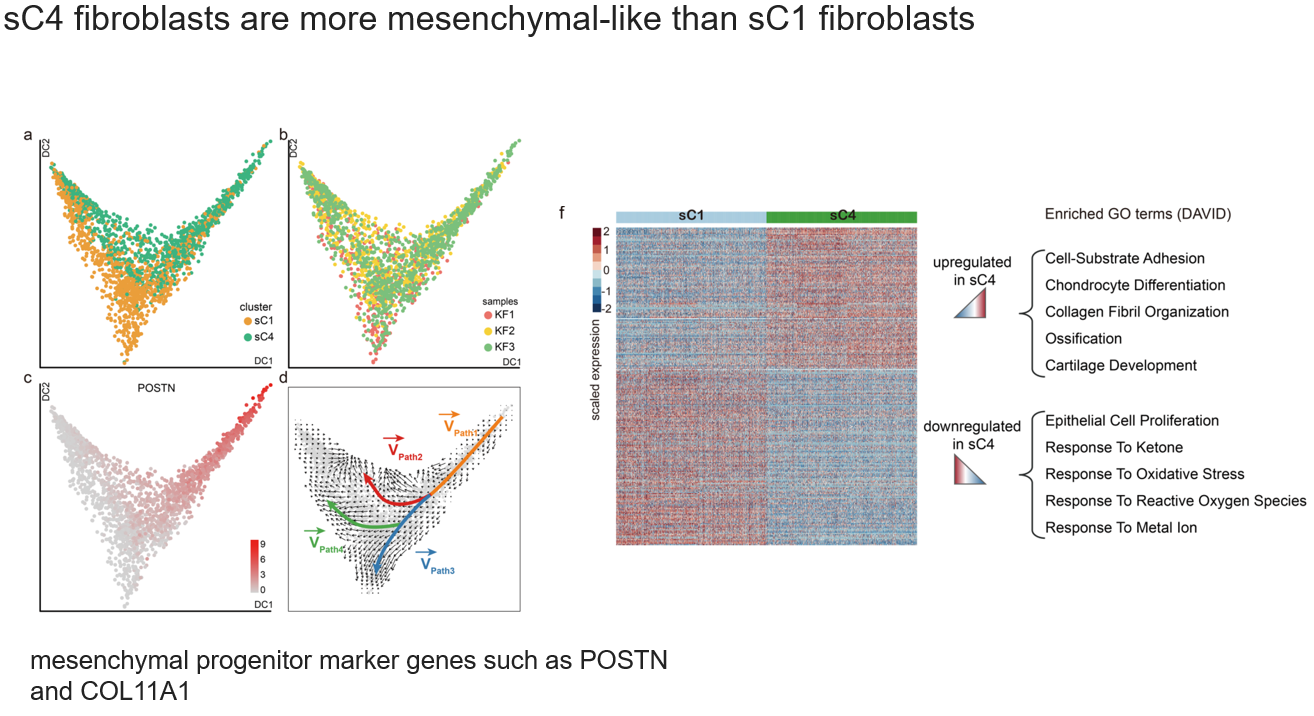
在3.2中,其实是把两个子类都归为MF了,这一部分就探究了它俩的关系。
- a, b. diffusion map,这里只能看出降维信息,两类并不重合
- c. mesenchymal progenitor marker genes在cS4中高表达(说明cS4可能是更早期的状态)
- d. RNA velocity分析给出了几条可能的转化路径
- f. 比较了cS4和cS1的差异,发现cS4更加具有间质特征
3.7 其他数据集也能发现这种MF细胞

- a. 别人的数据集(硬皮症,也是一种纤维化皮肤病)的降维注释结果;本研究中某个正常样本数据集的降维注释结果
- b, c. 提取上一步分析中两个数据集的成纤维细胞,整合,降维聚类
- d. MF的marker基因在降维图中的投影,发现在cluster 7中,这些marker高表达,且疾病组织比正常组织高表达
- e. cluster 7与本研究中4种成纤维细胞亚群的相关性热图,cluster 7和MF的相关性最高
结论是其他数据集也能找到这种MF,说明纤维化皮肤病存在一种广泛的致病机制
4. ref
Single-cell RNA-seq reveals fibroblast heterogeneity and increased mesenchymal fibroblasts in human fibrotic skin diseases
(2021.10.22组会分享的文献,因为准备时间有限,就选了这一篇较简单的)
我之前有想过一个问题,对于单细胞转录组数据,两组比较之后,如果已经找出有丰度差异的亚群,接下来的重点是:
①研究这一群的特征
还是
②比较这一群里面两组之间的差异
我当时是觉得①更好,因为把这种亚群的特征研究清楚了,反映的就是组间差异(富含这种亚群的condition,也具有更明显的这种亚群的特征)。另一方面②中的比较,因为细胞注释是相同的,这样做可能得不出什么差异或者差异很小(假设虽然有丰度差异,但都有一定的细胞量)。
这篇文献的前半部分cluster之间以及condition之间的比较都得出相似的结果,跟我之前想的有点不一样...
因水平有限,有错误的地方,欢迎批评指正!