来自知乎Live-孟浩巍
1.文章重要技术及图讲解
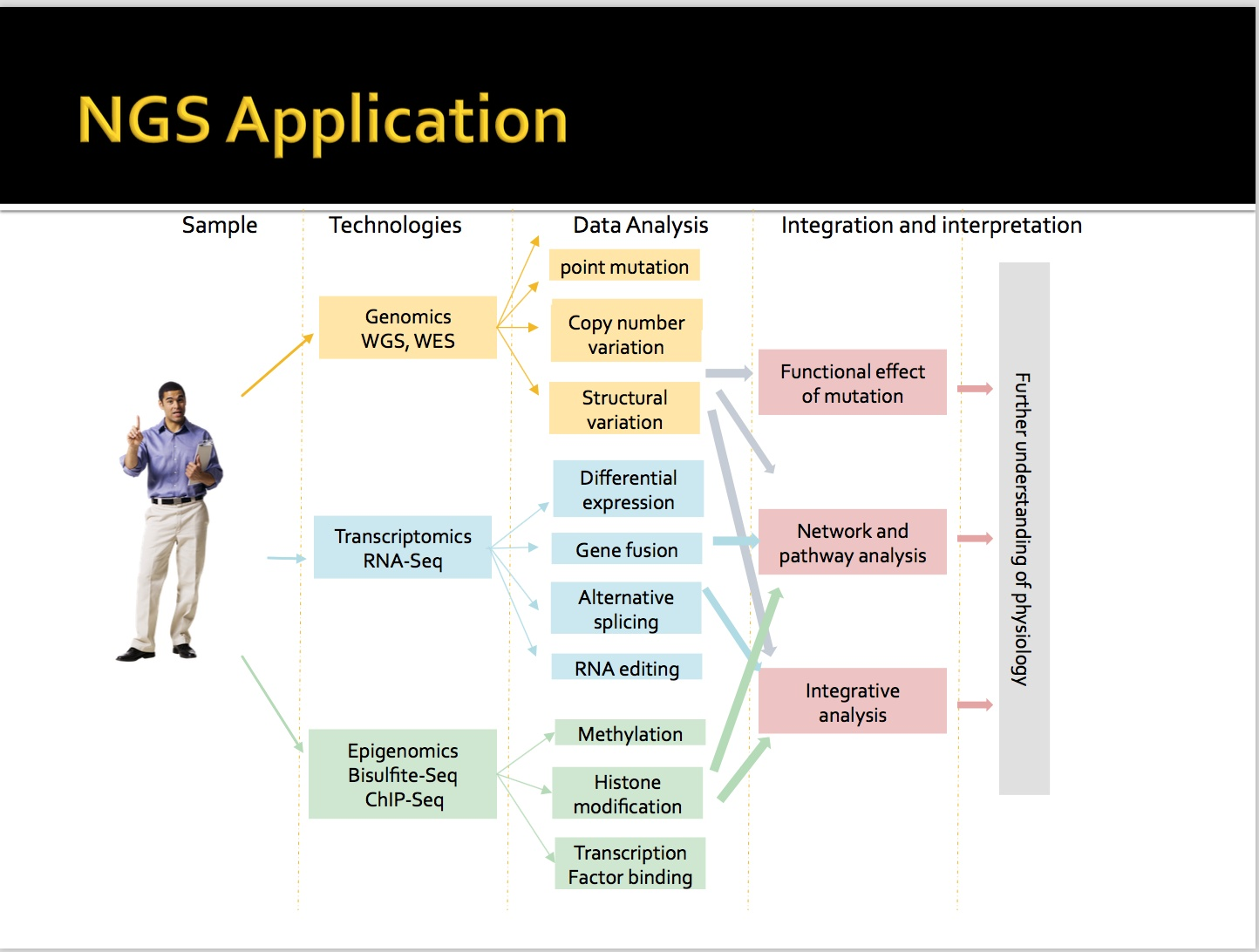
首先在转录组RNA-seq中,有基因表达差异、基因融合、可变剪切、RNA单点突变。
在基因组中,单点变异、结构变异,CNV变异(拷贝数变异)
三类基本内容,主要是RNA-seq的分析。
从生物水平角度理解肺癌,掌握RNA-seq的建库流程 ,单细胞的建库流程和方法。
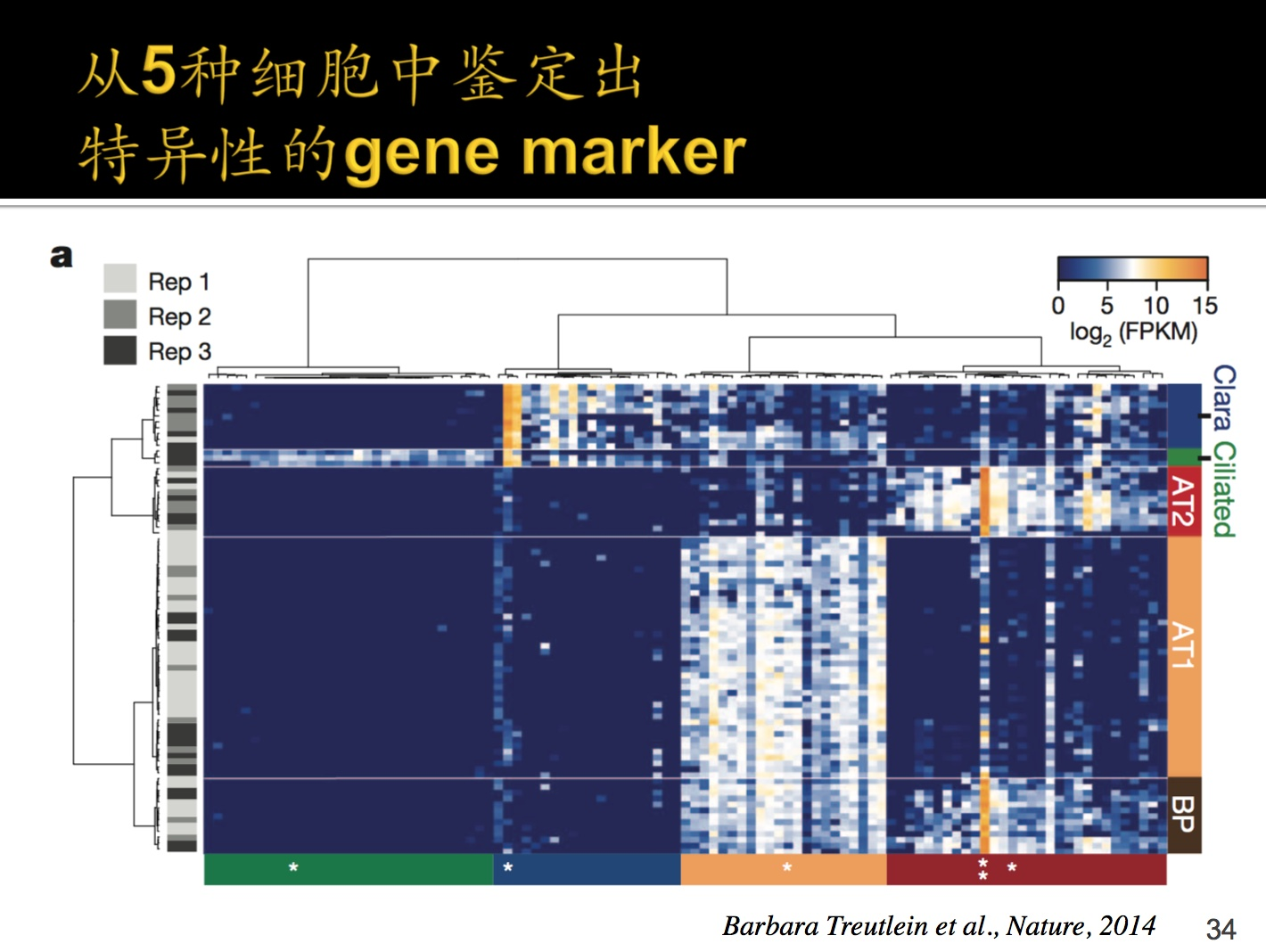
这个是通过热力图的方式,找出5种细胞中的特异性的gene marker,什么是基因标记,在某一个stage或者细胞内特异性表达的基因是基因marker。
0-15图标是取过log的fpkm,每一行是一个细胞的数据(约有80行),每一列一个基因的数据(有100+列)选取了100多个有代表性的基因。
左侧的rep1,2,3是聚类,是批次的意思。时间间隔大的叫做两个batch,同样的方法去做生物学重复叫repeat。每一个Repeat都有一个侧重点,根据颜色深度。颜色分布比较均匀说明不是因为聚类原因才导致的当前结果。
*是什么意思?表示相对应的细胞中的差异表达基因gene marker。

bp细胞可以分划为AT1和AT2细胞,导致基因表达谱逐渐发生变化,到6和4表达谱就完全不同了,包含了基因表达调控的过程。
灰色大括号:这个部分是两个Stage显著变化的基因。
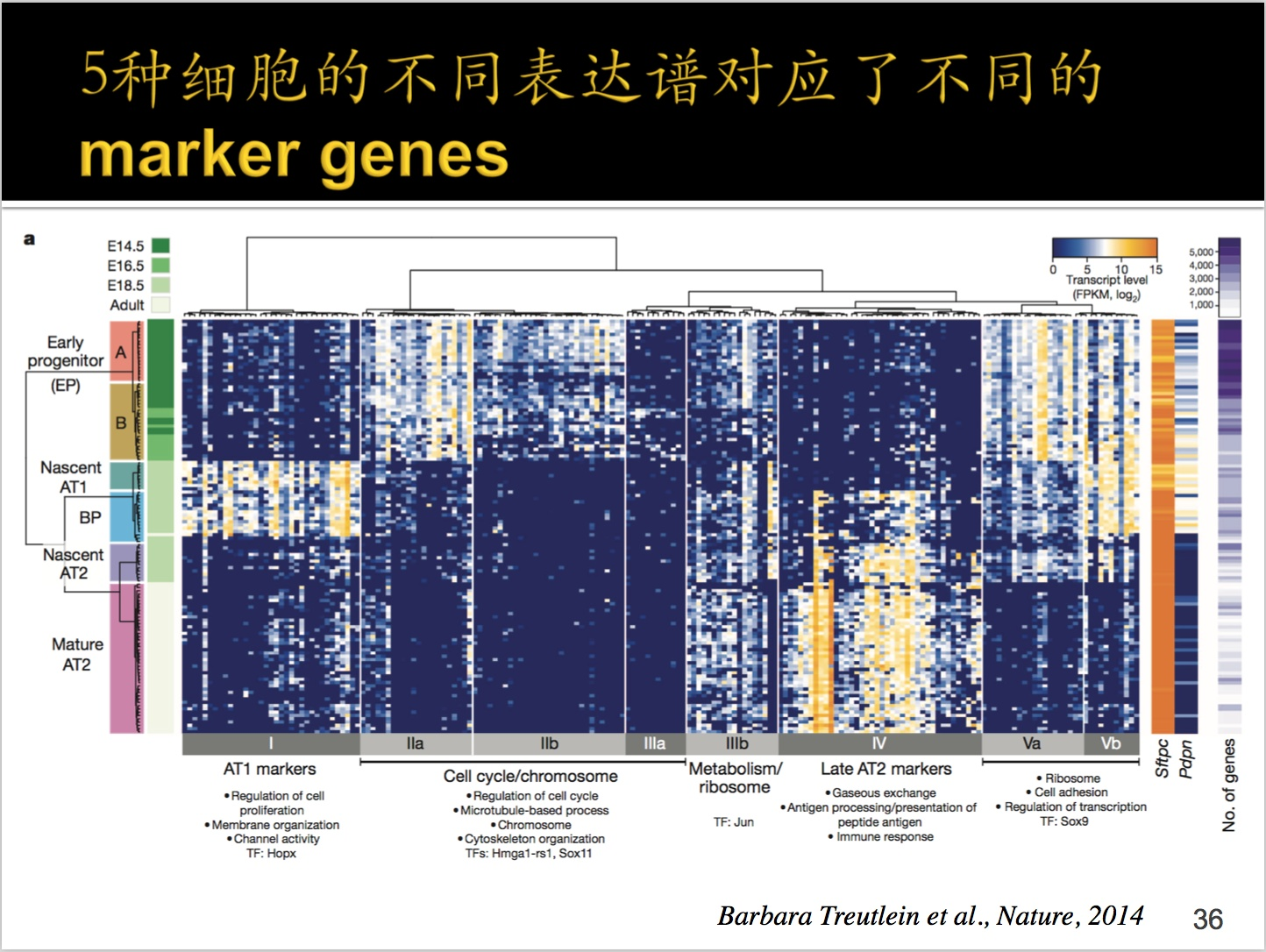
①左边绿色的部分,在胚胎发育过程当中的14.5天,16.5天到成熟时期分别进行了RNA-seq;
②右上角的基因fpkm基因表达量,越深表达量越低,越亮表达量越高;
③最左边的颜色部分是为了区分细胞。Early progenitor(早期祖先)。
④每一行是一个单细胞,每一列是一个基因。那么同一细胞之间是有聚类在一起的。调过参数、使用trick选择部分基因,才让BP出现在中间的,AT1和AT2是由BP发展而来的。
启示:能做出来这个结果,客观上真假不能确定,这种参数条件下能出来这种结果。
⑤横轴底部是将细胞分成不同的时期。
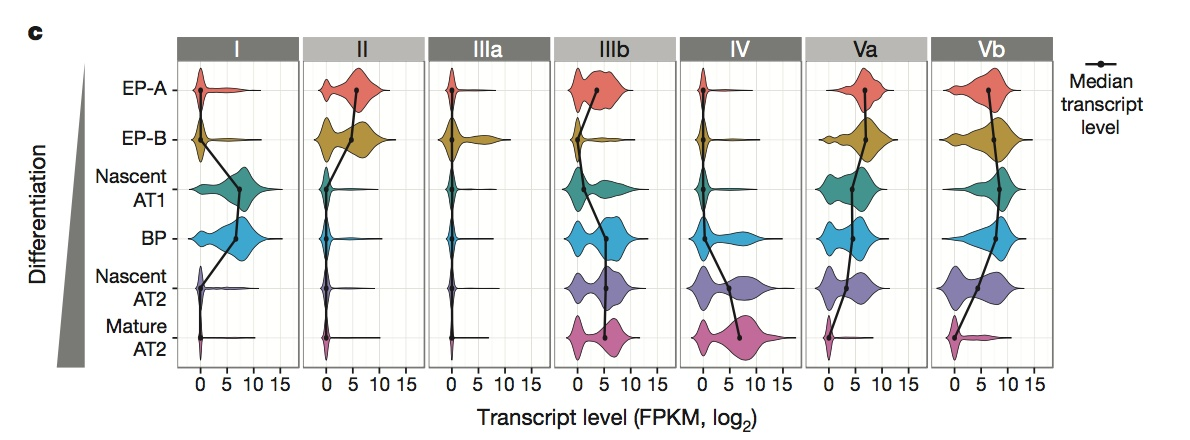
是boxplot的补充,随着分化的进行。随着分化的进行,纵向是不同的细胞类型,横轴是不同的基因list。
————使用RNA-seq,提供了分化表达时的基因marker,给出了每个过程较重要的基因list。

建库做了100+个细胞;PCA分析;花了很多的heatmap并且用到了聚类方法;用到了GO分析;画了whilelink???
2.具体技术细节

建库方法1:成熟的mRNA有5'端的帽子和3'端的PolyA尾巴,直接对PolyA富集,那么就可以针对成熟mRNA了,但是这样就扔掉了很多不成熟的。
建库方法2:rRNA minus去除法。
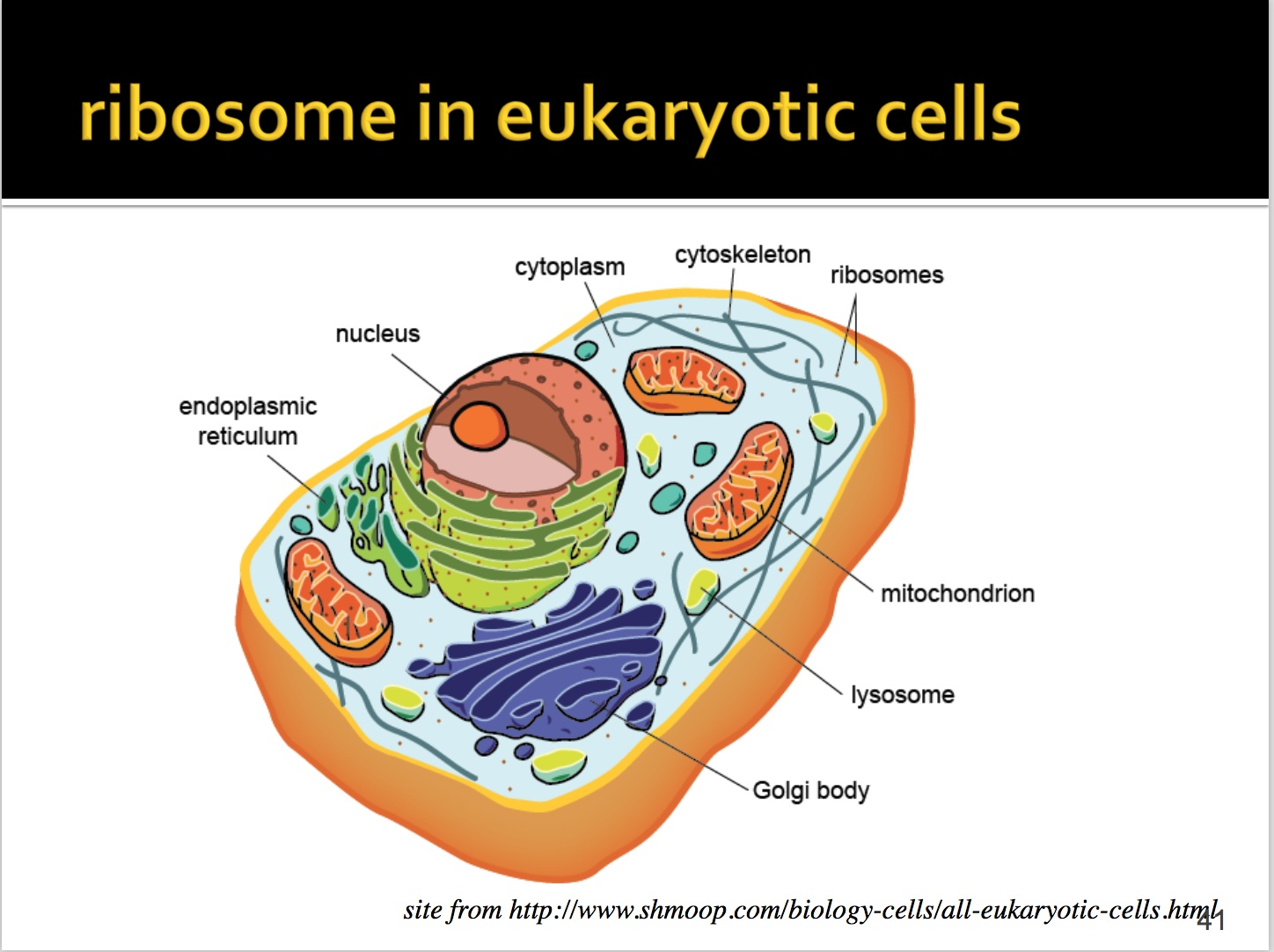
核糖体主要分布在两个位置:糙面内质网和游离态,主要是在内置网上。
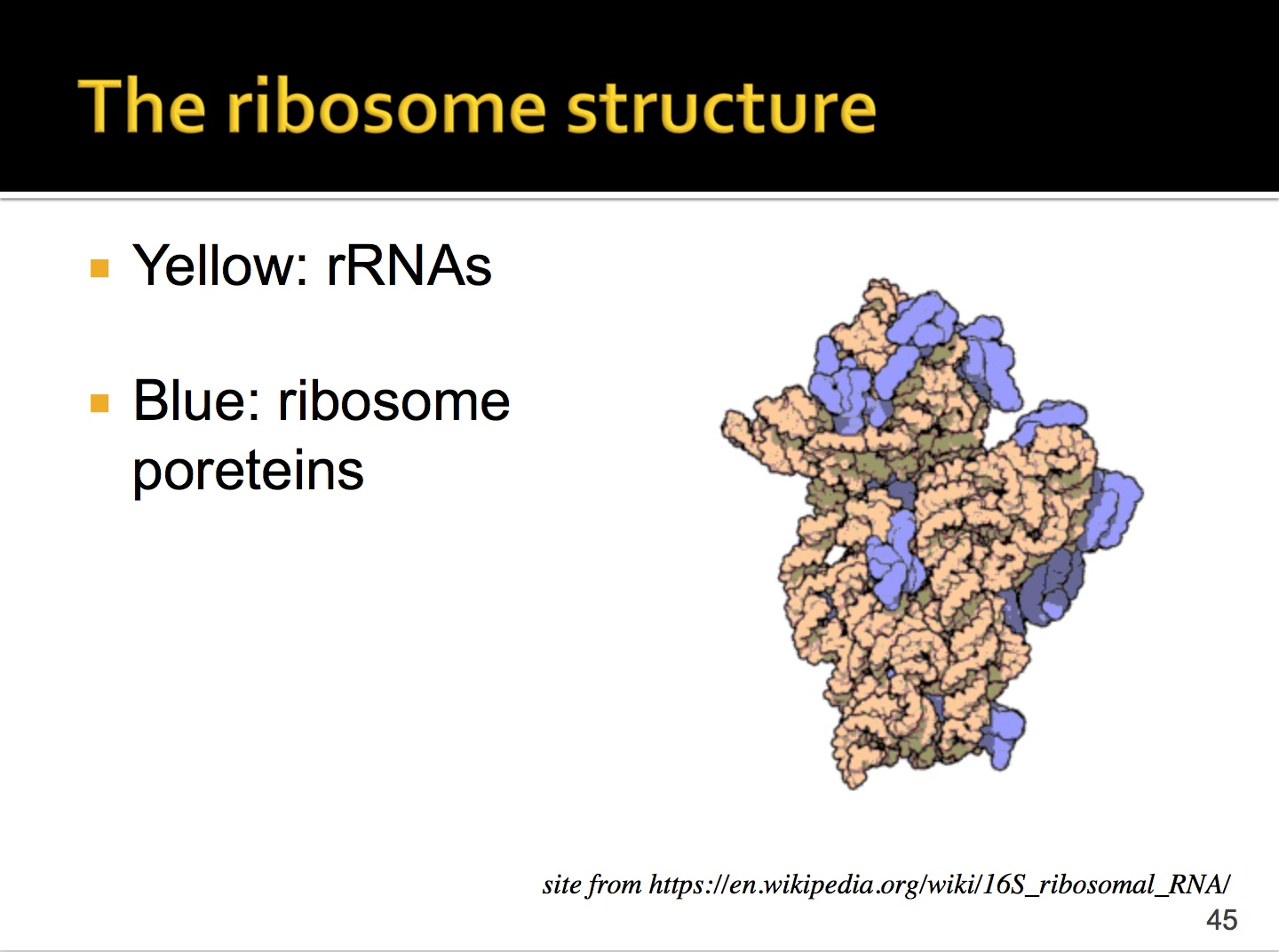
这个是核糖体的结构,黄色部分是rRNA,蓝色部分是核糖体蛋白。可以看出rRNA占了较大部分,核糖体蛋白主要是维持结构稳定。

//这个一开始看一脸懵,经过讲解看懂了。
左边是真核生物的rRNA,有一个大亚基和一个小亚基,其中大亚基是由5.8S,5S,28S这三种rRNA组成的,小亚基由18S rRNA组成的。
这四种亚基占细胞内RNA的比例99%以上,所以在建库时必须要去掉rRNA。
以上是部分关于建库的生物知识,下面是传统mRNA建库的正式的步骤:
Ⅰ.提取mRNA

1.使用Oligo dT磁珠对成熟的mRNA进行富集;
2.然后进行cDNA反转,就是根据mRNA得出互补的DNA基因片段;
3.打断成片段
4.加接头
5.进行PCR扩增
重点:3-4打断的过程,应该会不一般齐,有一个末端补平的过程,然后再加上一个A粘性末端,之后就可以加上测序的引物,引物是Y型的adaptor.
Ⅱ.rRNA minus方法
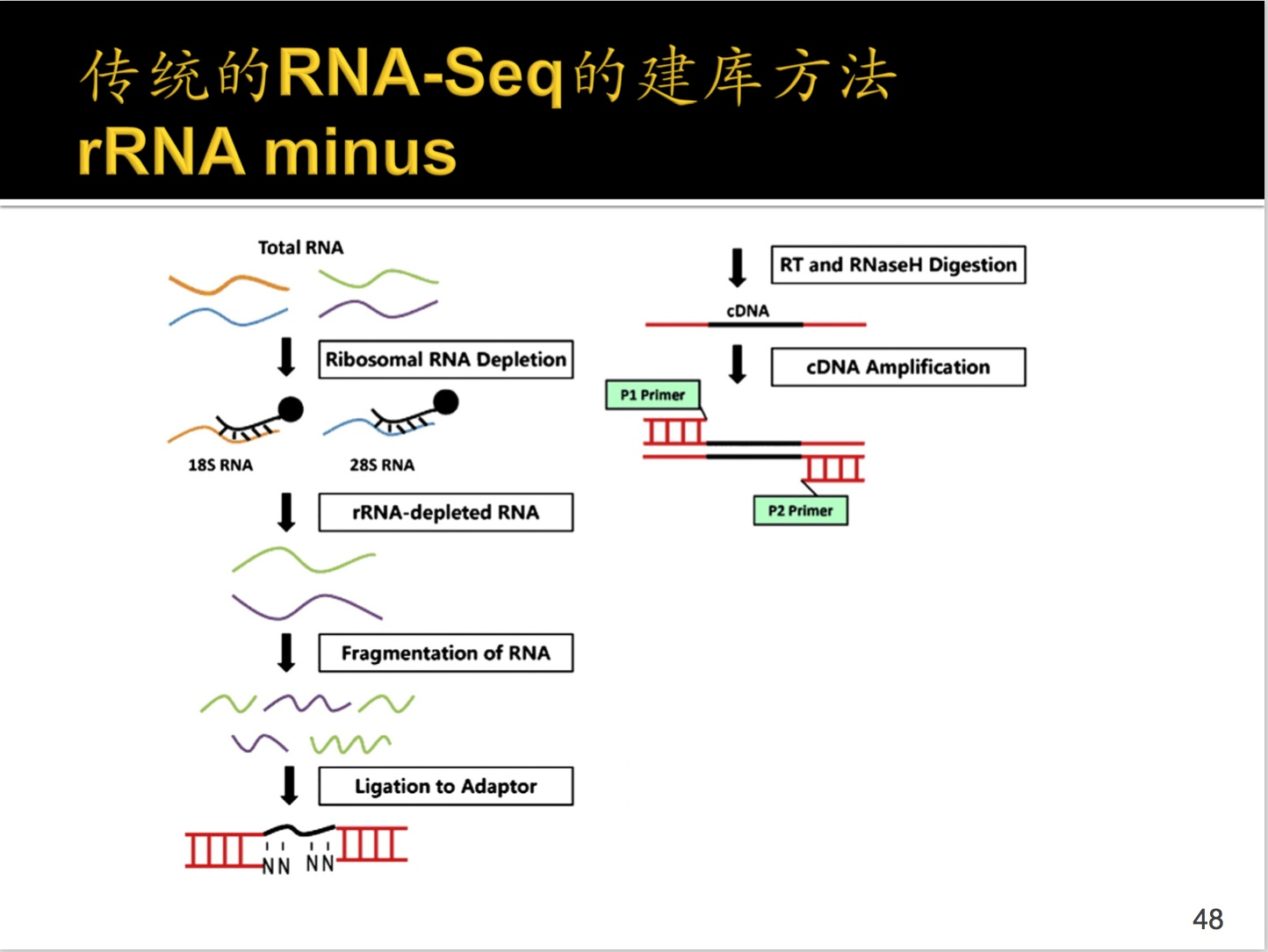
//这个听了两遍还是不太明白,是不是讲错了一点?
首先是将所有的RNA提取,然后是用磁珠将rRNA先去掉一遍,然后打碎,接上adaptor;转换成cDNA,如果此时还有rRNA,那么加入RNaseH进行消化掉,之后对加上引物cDNA进行扩增。
之后的过程就是一样了,这不就是建库完成了吗?~~~
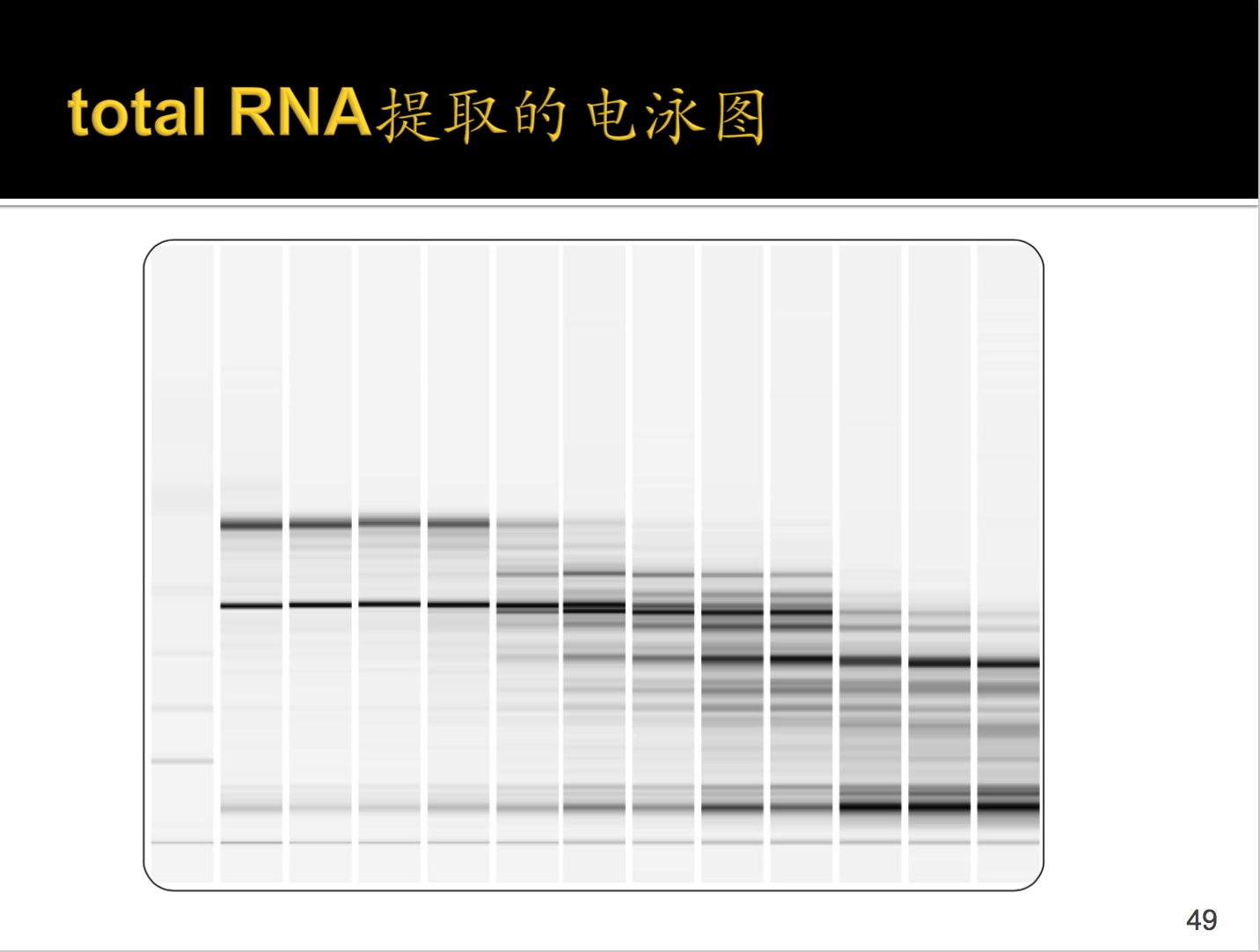
//很不明白这是什么意思,为什么要测基因表达差异呢?有什么作用呢???
那么如何判断提取的RNA的质量呢?通过电泳图,如上。
最左边是Gene marker,接着从左到右依次是从好到坏,最好的就是左边第二条,最差的是最右边的。
有人提问如何去 tRNA,回答是由于长度的选择会将tRNA筛掉。(我怎么就没想到这个问题呢?还是水平不行。)

这个图是一个量化RNA提取水平,RIN(RNA完整数),需要有这些region和一些fragment。
下面给出了几个标准:

RIN越大越好,提取组完全没有降解,可以进行下一步。一般要求RIN7以上,低于则建库失败。
有人提问:如何判断提取的RNA中有没有被别的物种污染?
答:这个是需要进行比对的,测序之后的GCcontent是可以看出来的,如果比对不上那么就BLAST。
有人提问:那个rRNA去除的时候那个磁珠是可以特异结合rRNA的?
答:是特异结合的,有两步的去rRNA的,第一步就是磁珠,第二步是加入rRNA降解酶消化掉。(为什么这种问题我没有想到?其实我这个地方是不太明白的,我的想法是以后有问题再百度,为什么我没有提出问题呢?。。。这就是差距,以及性格问题)
3.单细胞测序入门
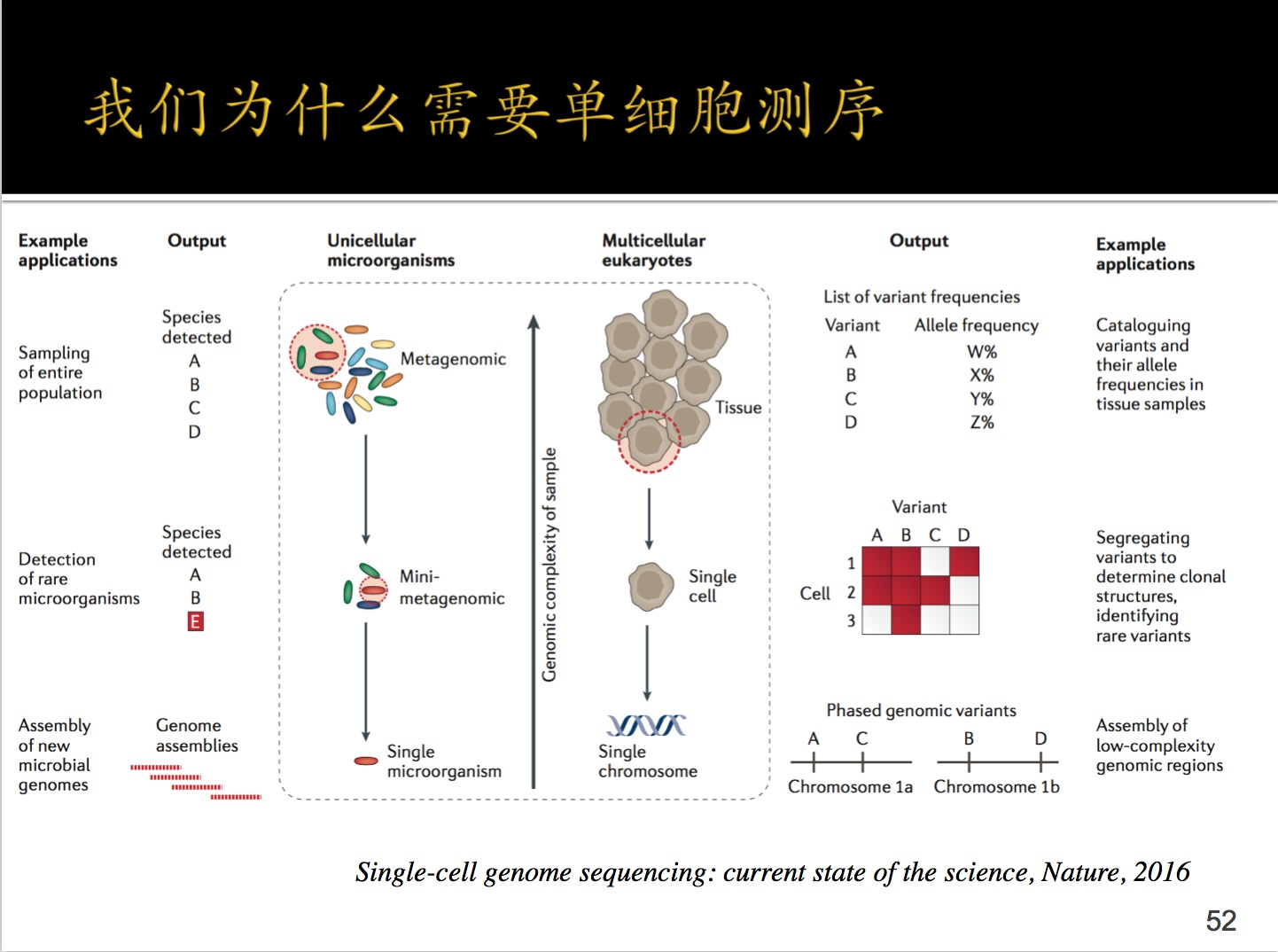
分为两种:DNA水平测序;RNA水平测序。
比如本篇文章的五种,肺部细胞最终形成了5种:科普一下:
小肺泡细胞(I型)、大肺泡细胞(II型肺泡细胞)、肺巨噬细胞等等类型。
这几种细胞的基因表达都不同,这就是单细胞存在的意义,就是基因差异性表达呗!
比如在上图中右部有一个红块,细胞1中ABD基因表达,细胞2中ABC基因表达....如果一块测那么所有肯定是都表达。
下面是09年的早期single-cell的Rna-seq老师提出的方法:

提问:大家可以思考一下,为什么传统的RNA-Seq不能做单细胞?
//我刚入门,感觉一脸懵,不知道。
只要有富集的过程,效率都是非常低的,能富集到10%就非常多了。总之就是富集效率低!
细胞裂解-> 带有T的primer直接进行cDNA反转-> 再加上polyA-> 再合成UP2-> 使用PCR扩增-> cDNA打断-> 绑定Adaptor-> 库扩增。
//为啥这里有一个cDNA的扩增呢?应该直接打断+adaptor,然后再PCR啊。
这里还是应该对cDNA有所学习:
与mRNA互补的DNA,是与RNA互补的单链DNA,在反转录酶的作用下合成的;合成完之后再在碱作用下去掉RNA,再合成双链cDNA,与原来基因中的DNA不同而且没有内含子!
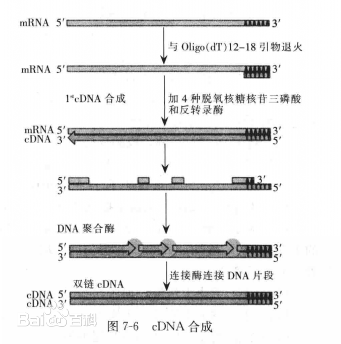
这个就是cDNA双链的合成过程,还是比较容易理解的!
**所有单细胞测序scRNA-seq的就是先反转再富集,一旦有富集就肯定做不到单细胞。
PCR扩增长度是有一定限制的(但是为什么为什么为什么?那里并没有打断就进行了PCR amplification呢???这不是相矛盾了吗???)
PCR扩增时有偏倚bias,它偏向于扩增GC含量高的。
下面是smart-seq的过程:
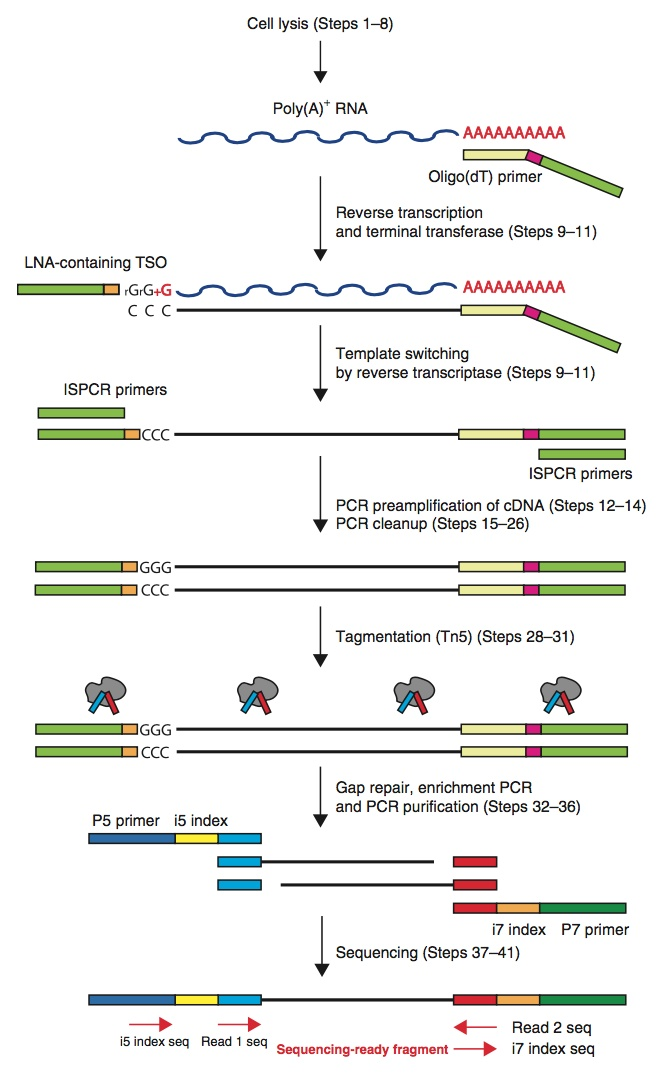
就是上边这个过程了,多加一个ployA,合成时加了一个CCC,之后用特殊酶Tn5直接会识别4个碱基并加上Adaptor,
接下来的过程和RNA-seq还是差不多的,首先是质控,mRNA前处理,回帖(也就是比对),计算不同基因表达量,比较差异表达。
有个人提问:全程没有rRNA去除的步骤,是不是pcr扩增mRNA之后rRNA被稀释了?(我为什么没想到,之前你想想建库的时候不就是苦于rRNA过多吗???)
答:+adaptor其他的序列是加不上的,所以建库是建不了rRNA的。(感觉live主这个回答的有点水,不具体)
//有可能是只有有一个polyA的才能再加一个polyA ???我是这么理解。