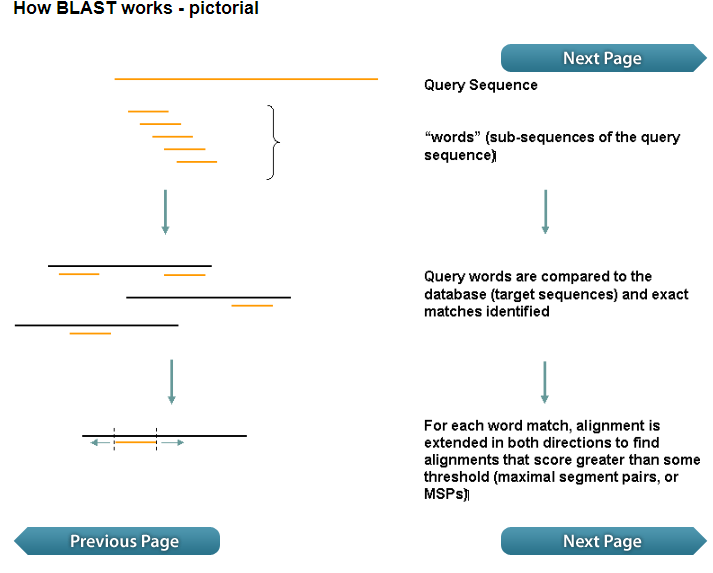
blast的原理就是将想要明确注释的sequence(这个sequence就是query)先打断,即一条sequence变成多条sub-sequence(sub-sequence也就是word),然后拿这些sub-sequence与数据库中的序列比较(数据库中的序列是已经注释过的),然后将这些word向两边延展,延展方式是将单个word(就是图中黄色的线)对应的sequence(就是图中黑色的线)保持不变,拿其他word的信息mapping到黑色线上。
联系到实际实验就是,我手头的pep文件中的34条蛋白质序列就是34个query(就是黄色线),在blast中,先将这34条序列每一条都打断,然后与斑马雀.fa的数据库(就是黑色线)相互匹配,所以得到的结果是某一条scaffold(黑色线)与某一个gene(黄色线)的匹配情况。目的是想知道在某个物种中,某些gene的分布情况。
由此产生的gff文件中的内容是:
1.scaf_id
2.gene_name
3.scaf_len(整个一条sacffold的length)
4.sacf_start(可以mapping到这个gene_name的word的起始位点)
5.scaf_end(可以mapping到这个gene_name的word的终止位点,scaf_start与scaf_end之间的内容包括了exon和intron)
6.block_number(是scaf_start与scaf_end之间的gene_name的exon个数)
7.block_start(某个block的起始位点,此处可能有多个,如果block_number是3,则有3个block,也就有3个block_start)
8.block_end(某个block的终止位点,此处可能有多个,如果block_number是3,则有3个block,也就有3个block_end)
9.identify(某个block的identify,此处可能有多个,如果block_number是3,则有3个block,也就有3个identify)
10.align_rate