全基因组测序
全基因组测序分为从头测序(de novo sequencing)和重测序(re-sequencing)。
从头测序(de novo)不需要任何参考基因组信息即可对某个物种的基因组进行测序,利用生物信息学分析方法进行拼接、组装,获得该物种的基因组序列图谱,从而推进该物种的后续研究。
基因组重测序 是对有参考基因组物种的不同个体进行的基因组测序,并在此基础上对个体或群体进行差异性分析。
基因组重测序主要用于辅助研究者发现单核苷酸多态性位点(SNPs)、拷贝数变异(CNV)、插入/缺失(Indel)等变异类型,以较低的价格将单个参考基因组信息扩增为生物群体的遗传特征。全基因组重测序在人类疾病和动植物育种研究中广泛应用。
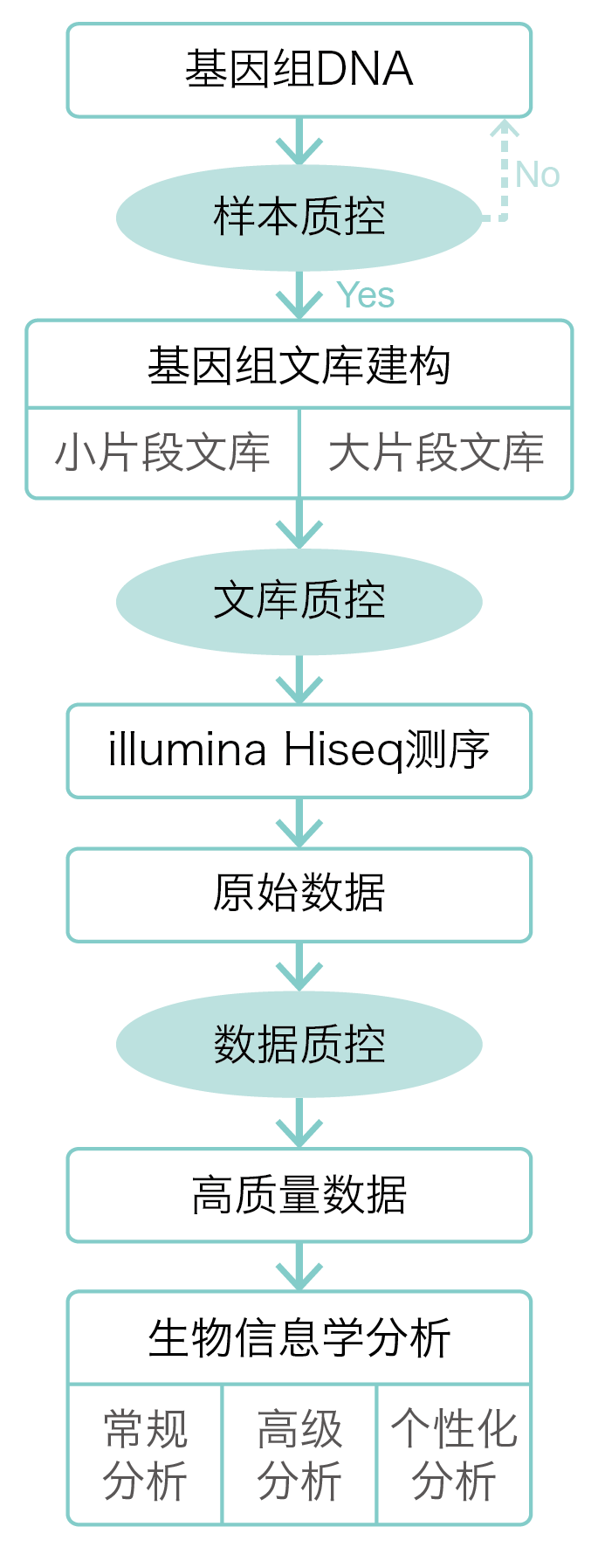
技术路线
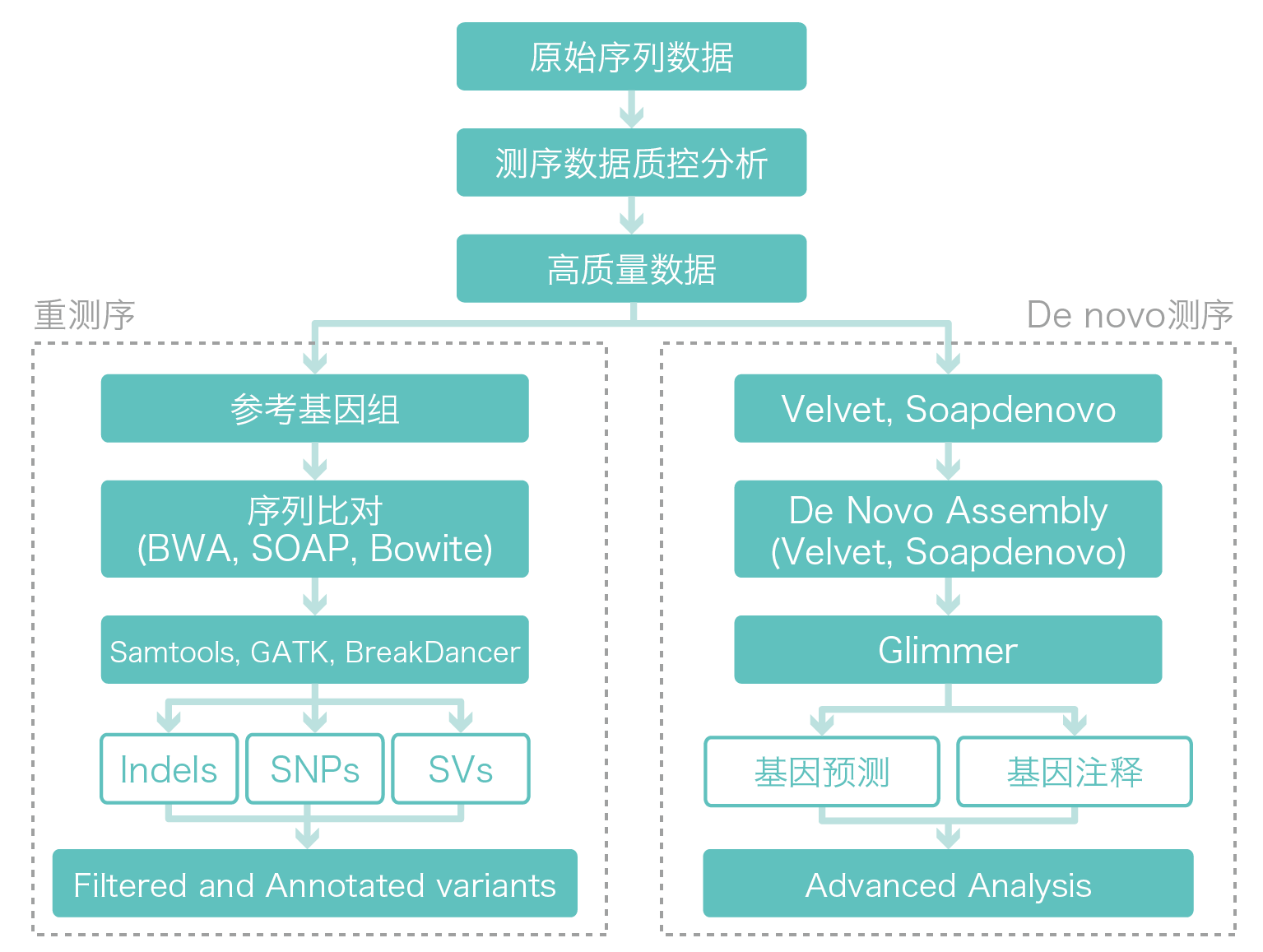
生物信息分析

案例解析
1.比较基因组分析
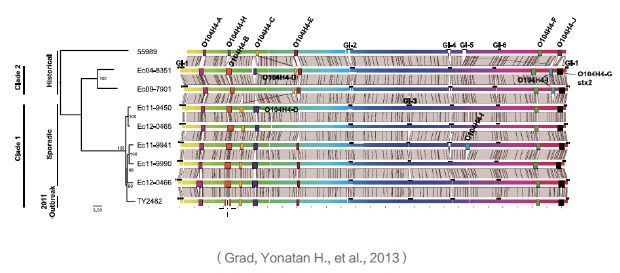
采用progressiveMauve软件比对9株大肠杆菌O104:H4分离株的染色体序列,展示可移动遗传元件和基因组可变区域信息,利用核心SNP位点信息构建最大似然进化树揭示菌株间的亲缘关系。
2.重复序列分析
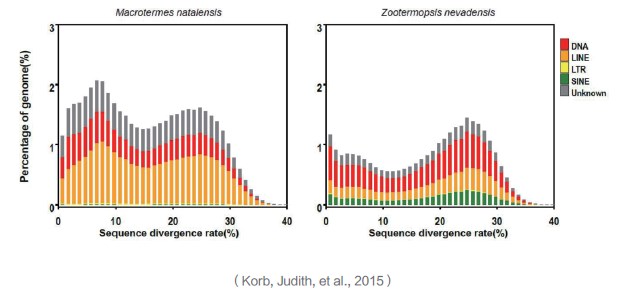
采用从头预测和基于数据库比对的两种方法对纳塔尔大白蚁和湿木白蚁的基因组序列进行转座子(TEs)分析,利用RepeatModeler软件对两种方法的结果进行整合分析并构建转座子序列数据库,使用RepeatClassifier软件对转座子进行分类,计算两种白蚁基因组中转座子的序列变异速率,揭示基因组扩张的可能机制。
3.代谢通路重建
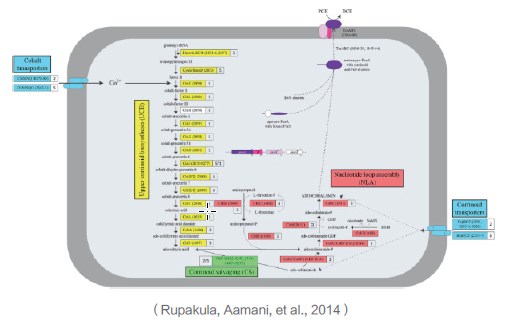
根据限制性脱氯细菌(PER-K23)基因组注释信息,预测类咕啉的生物合成包含4种代谢途径。
4.基因进化分析
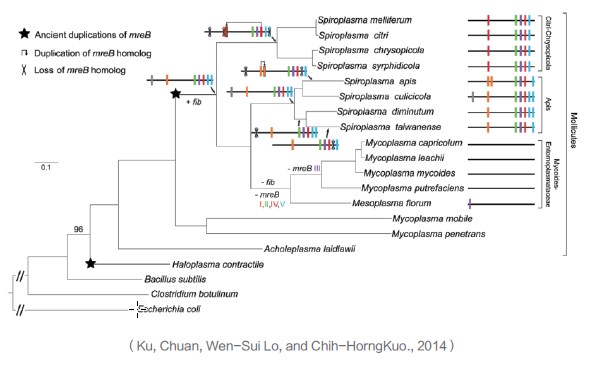
利用117个单拷贝编码蛋白的基因序列构建Mollicutes、Haloplasma和Firmicutes菌株的最大似然物种进化树,揭示不同菌株基因组中mreB和fib基因的获得与丢失。
测序策略及数据量
测序策略:PE125或PE150
建议数据量:根据基因组大小进行30×或50×的测序
建议数据量:根据基因组大小进行30×或50×的测序