Opportunities and challenges in long-read sequencing data analysis 长时间测序数据分析的机遇和挑战
Abstract
Long-read technologies are overcoming early limitations in accuracy and throughput, broadening their application domains in genomics. Dedicated analysis tools that take into account the characteristics of long-read data are thus required, but the fast pace of development of such tools can be overwhelming. To assist in the design and analysis of long-read sequencing projects, we review the current landscape of available tools and present an online interactive database, long-read-tools.org, to facilitate their browsing. We further focus on the principles of error correction, base modification detection, and long-read transcriptomics analysis and highlight the challenges that remain.
摘要
长read测序技术在准确性和通量方面克服了早期的局限性,拓宽了其在基因组学中的应用领域。 因此,需要专门的分析工具来考虑长read的数据的特征,但是这种工具的开发速度可能会非常快。 为了帮助设计和分析长read的排序项目,我们回顾了可用工具的现状,并提供了一个在线交互式数据库,long-read-tools.org,以方便他们浏览。 我们进一步关注纠错、基修改检测和长read的转录组分析的原则,并强调仍然存在的挑战。
关键词:长读测序,数据分析,PacBio,牛津纳米孔
Introduction
Long-read sequencing, or third-generation sequencing, offers a number of advantages over short-read sequencing [1, 2]. While short-read sequencers such as Illumina’s NovaSeq, HiSeq, NextSeq, and MiSeq instruments [3–5]; BGI’s MGISEQ and BGISEQ models [6]; or Thermo Fisher’s Ion Torrent sequencers [7, 8] produce reads of up to 600 bases, long-read sequencing technologies routinely generate reads in excess of 10 kb [1].
Short-read sequencing is cost-effective, accurate, and supported by a wide range of analysis tools and pipelines [9]. However, natural nucleic acid polymers span eight orders of magnitude in length, and sequencing them in short amplified fragments complicates the task of reconstructing and counting the original molecules. Long reads can thus improve de novo assembly, mapping certainty, transcript isoform identification, and detection of structural variants. Furthermore, long-read sequencing of native molecules, both DNA and RNA, eliminates amplification bias while preserving base modifications [10]. These capabilities, together with continuing progress in accuracy, throughput, and cost reduction, have begun to make long-read sequencing an option for a broad range of applications in genomics for model and non-model organisms [2, 11].
Two technologies currently dominate the long-read sequencing space: Pacific Biosciences’ (PacBio) single-molecule real-time (SMRT) sequencing and Oxford Nanopore Technologies’ (ONT) nanopore sequencing. We henceforth refer to these simply as SMRT and nanopore sequencing. SMRT and nanopore sequencing technologies were commercially released in 2011 and 2014, respectively, and since then have become suitable for an increasing number of applications. The data that these platforms produce differ qualitatively from second-generation sequencing, thus necessitating tailored analysis tools.
Given the broadening interest in long-read sequencing and the fast-paced development of applications and tools, the current review aims to provide a description of the guiding principles of long-read data analysis, a survey of the available tools for different tasks as well as a discussion of the areas in long-read analysis that require improvements. We also introduce the complementary open-source catalogue of long-read analysis tools: long-read-tools.org. The long-read-tools.org database allows users to search and filter tools based on various parameters such as technology or application.
介绍
与短读测序相比,长读测序或第三代测序具有许多优势[1,2]。 而短读测序仪,如Illumina的NovaSeq、HiSeq、NextSeq和MiSeq仪器[3-5]; BGI MGISEQ和BGISEQ机型[6];或者Thermo Fisher的离子激流测序仪[7,8]可以产生多达600个碱基的序列, 长读测序技术通常可以产生超过10kb的[1]序列。
短读测序是经济有效的,准确的,并支持广泛的分析工具和管道[9]。 然而,天然核酸聚合物的长度跨度为8个数量级,对它们进行简短的扩增片段测序,使得重建和计算原始分子 的任务变得更加复杂。 长read因此可以提高从头组装、绘图确定性、转录亚型识别和结构变异的检测。 此外,对天然分子(包括DNA和RNA)进行长时间测序,消除了扩增偏倚,同时保留了碱基修饰[10]。 这些能力,加上在准确性、通量和成本降低方面的持续进步, 已经开始使长读测序成为模型和非模型生物基因组学中广泛应用的选择[2,11]。
目前有两种技术在长时间测序领域占据主导地位: 太平洋生物科学公司(PacBio)的单分子实时测序(SMRT)和牛津纳米孔技术公司(ONT)的纳米孔测序。 我们将其简单地称为SMRT和纳米孔测序。 SMRT和纳米孔测序技术分别于2011年和2014年商业化发布,自那以后,它们的应用越来越广泛。 这些平台的数据从第二代测序产生定性不同,因此需要定制的分析工具。 鉴于拓宽兴趣读排序应用程序和工具的快速发展,当前审查旨在提供一个描述的读数据分析的指导原则, 一项调查显示可用的工具,不同的任务以及讨论读区域的分析,需要改进。 我们还介绍了几个 长read分析工具 的互补开源读目录 :long-read-tools.org。 long-read-tools.org允许用户根据各种参数(如技术或应用程序)搜索和过滤工具。
The state of long-read sequencing and data analysis
Nanopore and SMRT long-read sequencing technologies rely on very distinct principles. Nanopore sequencers (MinION, GridION, and PromethION) measure the ionic current fluctuations when single-stranded nucleic acids pass through biological nanopores [12, 13]. Different nucleotides confer different resistances to the stretch of nucleic acid within the pore; therefore, the sequence of bases can be inferred from the specific patterns of current variation. SMRT sequencers (RSII, Sequel, and Sequel II) detect fluorescence events that correspond to the addition of one specific nucleotide by a polymerase tethered to the bottom of a tiny well [14, 15].
Read length in SMRT sequencing is limited by the longevity of the polymerase. A faster polymerase for the Sequel sequencer introduced with chemistry v3 in 2018 increased the read lengths to an average 30-kb polymerase read length. The library insert sizes amenable to SMRT sequencing range from 250 bp to 50 kbp. Nanopore sequencing provides the longest read lengths, from 500 bp to the current record of 2.3 Mb [16], with 10–30-kb genomic libraries being common. Read length in nanopore sequencing is mostly limited by the ability to deliver very high-molecular weight DNA to the pore and the negative impact this has on run yield [17]. Basecalling accuracy of reads produced by both these technologies have dramatically increased in the recent past, and the raw base-called error rate is claimed to have been reduced to < 1% for SMRT sequencers [18] and < 5% for nanopore sequences [17].
While nanopore and SMRT are true long-read sequencing technologies and the focus of this review, there are also synthetic long-read sequencing approaches. These include linked reads, proximity ligation strategies, and optical mapping [19–28], which can be employed in synergy with true long reads.
With the potential for accurately assembling and re-assembling genomes [17, 29–32], methylomes [33, 34], variants [18], isoforms [35, 36], haplotypes [37–39], or species [40, 41], tools to analyse the sequencing data provided by long-read sequencing platforms are being actively developed, especially since 2011 (Fig. 1a).
纳米孔和SMRT长读测序技术依赖于非常独特的原则。
纳米孔测序仪(MinION、GridION和PromethION)测量单链核酸通过生物纳米孔时离子电流的波动[12,13]。
不同的核苷酸对孔内核酸的伸展具有不同的抗性;
因此,根据电流变化的具体规律可以推断出碱基的序列。
SMRT测序仪(RSII, Sequel,和Sequel II)检测荧光事件,这些荧光事件对应于拴在小孔底部的聚合酶添加的一个特定的核苷酸[14,15]。
SMRT测序中的阅读长度受到聚合酶寿命的限制。
2018年在《化学》v3中引入的更快的聚合酶将续篇测序器的读取长度增加到平均30 kb的聚合酶读取长度。
适合SMRT测序的库插入大小从250 bp到50 kbp不等。
纳米孔测序提供了最长的读取长度,从500 bp到目前记录的2.3 Mb[16],通常有10 - 30 kb的基因组文库。
纳米孔测序的读取长度主要受到将非常高分子量的DNA传输到孔的能力以及对运行产额[17]的负面影响的限制。
在过去的一段时间里,这两种技术所产生读取的碱基测定精度都有了显著提高,而SMRT测序器[18]和纳米孔序列[17]的原始碱基被称为错误率被降低到1%。
虽然纳米孔和SMRT是真正的长read测序技术,也是本综述的重点,但也有合成的长时间测序方法。
这些方法包括链读、近距离连接策略和光学映射[19-28],这些方法可以与真正的长读协同使用。
特别是自2011年以来,由于有可能精确组装和重新组装基因组[17,29 - 32]、甲基体[33,34]、变异体[18]、异构体[35,36]、单倍型[37-39]或物种[40,41],分析由长读测序平台提供的测序数据的工具正在积极开发(图1a)。
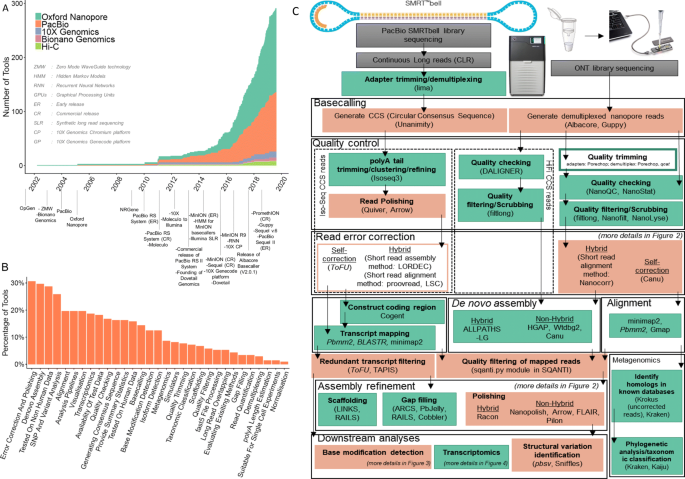
Overview of long-read analysis tools and pipelines.
a Release of tools identified from various sources and milestones of long-read sequencing.
b Functional categories.
c Typical long-read analysis pipelines for SMRT and nanopore data.
Six main stages are identified through the presented workflow (i.e. basecalling, quality control, read error correction, assembly/alignment, assembly refinement, and downstream analyses). The green-coloured boxes represent processes common to both short-read and long-read analyses. The orange-coloured boxes represent the processes unique to long-read analyses. Unfilled boxes represent optional steps.
Commonly used tools for each step in long-read analysis are within brackets.
Italics signify tools developed by either PacBio or ONT companies, and non-italics signify tools developed by external parties.
Arrows represent the direction of the workflow
长read的分析工具和管道概述。
a. 发布从各种来源和里程碑的长read排序确定的工具。
b 功能类别。
c 典型的SMRT和纳米孔数据长读分析管道。
通过目前的工作流程确定了六个主要阶段(即基础设置、质量控制、读取错误纠正、装配/对准、装配改进和下游分析)。
绿色的框表示短读和长读分析的共同过程。
橙色的盒子代表了长时间阅读分析的独特过程。
未填充的框表示可选步骤。
用于长时间分析的每个步骤的常用工具都在括号内。
斜体表示PacBio或公司,开发的工具和
non-italics表示工具开发的外部各方。
箭头表示工作流的方向
A search through publications, preprints, online repositories, and social media identified 354 long-read analysis tools. The majority of these tools are developed for nanopore read analyses (262) while there are 170 tools developed to analyse SMRT data (Fig. 1a). We categorised them into 31 groups based on their functionality (Fig. 1b). This identified trends in the evolution of research interests: likely due to the modest initial throughput of long-read sequencing technologies, the majority of tools were tested on non-human data; tools for de novo assembly, error correction, and polishing categories have received the most attention, while transcriptome analysis is still in early stages of development (Fig. 1b).
We present an overview of the analysis pipelines for nanopore and SMRT data and highlight popular tools (Fig. 1c). We do not attempt to provide a comprehensive review of tool performance for all long-read applications; dedicated benchmark studies are irreplaceable, and we refer our readers to those when possible. Instead, we present the principles and potential pitfalls of long-read data analysis with a focus on some of the main types of downstream analyses: structural variant calling, error correction, detection of base modifications, and transcriptomics.
通过出版物、预印本、在线存储库和社交媒体的搜索,我们发现了354种读过很久的分析工具。
这些工具大多数用于纳米孔读取分析(262),而有170种工具用于分析SMRT数据(图1a)。
我们根据它们的功能将它们分为31组(图1b)。
这确定了研究兴趣的发展趋势:可能是由于长读测序技术的初始通量不大,大多数工具都是在非人类数据上测试的;
用于重新装配、纠错和分类抛光的工具受到了最多的关注,而转录组分析仍处于发展的早期阶段(图1b)。
我们概述了纳米孔和SMRT数据的分析管道,并强调了流行的工具(图1c)。
我们不试图为所有长read的应用提供工具性能的全面审查;
专门的基准研究是不可替代的,我们会在可能的情况下推荐我们的读者。
相反,我们展示了长时间阅读数据分析的原则和潜在的陷阱,重点关注一些主要类型的下游分析:结构变体调用、错误纠正、检测碱基修改和转录组。
Basecalling
The first step in any long-read analysis is basecalling, or the conversion from raw data to nucleic acid sequences (Fig. 1c). This step receives greater attention for long reads than short reads where it is more standardised and usually performed using proprietary software. Nanopore basecalling is itself more complex than SMRT basecalling, and more options are available: of the 26 tools related to basecalling that we identified, 23 relate to nanopore sequencing.
During SMRT sequencing, successions of fluorescence flashes are recorded as a movie. Because the template is circular, the polymerase may go over both strands of the DNA fragment multiple times. SMRT basecalling starts with segmenting the fluorescence trace into pulses and converting the pulses into bases, resulting in a continuous long read (also called polymerase read). This read is then split into subreads, where each subread corresponds to 1 pass over the library insert, without the linker sequences. Subreads are stored as an unaligned BAM file. From aligning these subreads together, an accurate consensus circular sequence (CCS) for the insert is derived [42]. SMRT basecallers are chiefly developed internally and require training specific to the chemistry version used. The current basecalling workflow is ccs [43].
Nanopore raw data are current intensity values measured at 4 kHz saved in fast5 format, built on HDF5. Basecalling of nanopore reads is an area of active research, where algorithms are quickly evolving (neural networks have supplanted HMMs, and various neural networks structures are being tested [44]) as are the chemistries for which they are trained. ONT makes available a production basecaller (Guppy, currently) as well as development versions (Flappie, Scrappie, Taiyaki, Runnie, and Bonito) [45]. Generally, the production basecaller provides the best accuracy and most stable performance and is suitable for most users [46]. Development basecallers can be used to test features, for example, homopolymer accuracy, variant detection, or base modification detection, but they are not necessarily optimised for speed or overall accuracy. In time, improvements make their way into the production basecaller. For example, Scrappie currently maps homopolymers explicitly [47].
Independent basecaller with different network structures are also available, most prominently Chiron [48]. These have been reviewed and their performance evaluated elsewhere [13, 46, 49]. The ability to train one’s own basecalling model opens the possibility to improve basecalling performance by tailoring the model to the sample’s characteristics [46]. As a corollary, users have to keep in mind that the effective accuracy of the basecaller on their data set may be lower than the advertised accuracy. For example, ONT’s basecallers are currently trained on a mixture of human, yeast, and bacterial DNA; their performance on plant DNA where non-CG methylation is abundant may be lower [50]. As the very regular updates to the production Guppy basecaller testify, basecalling remains an active area of development.
碱基判定
任何长时间读取的分析的第一步都是建立碱基,即从原始数据到核酸序列的转换(图1c)。
这一步受到更大的注意长阅读比短阅读,它是更标准化的,通常执行专有软件。
Nanopore basecalling(碱基判定)本身比SMRT basecalling(碱基判定)更复杂,而且有更多的选择:在我们确定的26种与 basecalling(碱基判定)相关的工具中,有23种与Nanopore测序相关。
在SMRT测序过程中,荧光闪烁的序列被记录为电影。
因为模板是循环的,所以聚合酶可能会多次穿过DNA片段的两条链。
SMRT的基础检测开始于将荧光信号分割成脉冲并将脉冲转换成碱基,从而产生连续的长读(也称为聚合酶读)。
然后,这个读操作被拆分为子读操作,每个子读操作都对应于库插入的1次传递,没有链接器序列。
子读取被存储为一个未对齐的BAM文件。
通过将这些子读对齐在一起,就可以得到用于插入的精确的一致循环序列[42]。
SMRT基础测试人员主要是在内部开发的,需要针对所使用的化学版本进行专门的培训。
当前的基础工作流程是ccs[43]。
纳米孔原始数据是以4khz测量的当前强度值,以fast5格式保存,建立在HDF5上。
纳米孔读取的碱基化是一个活跃的研究领域,算法正在迅速发展(神经网络已经取代HMMs,各种神经网络结构正在[44]测试),它们所训练的化学反应也是如此。
ONT提供了一个生产基本版本(目前的Guppy)以及开发版本(Flappie, Scrappie, Taiyaki, Runnie,和鲣鱼)[45]。
一般情况下,生产的基管提供了最好的精度和最稳定的性能,适用于大多数[46]用户。
开发基测试器可用于测试特性,例如,均聚物精度、变体检测或基修改检测,但它们不一定优化速度或整体精度。
随着时间的推移,改进工作进入了生产基础设施。
例如,Scrappie目前显式地映射均聚物[47]。
具有不同网络结构的独立基包也可用,最突出的是Chiron[48]。
这些已经在其他地方进行了审查和绩效评估[13,46,49]。
训练自己的 basecalling(碱基判定)模型的能力打开了改进 basecalling(碱基判定)性能的可能性,方法是根据样本的特征[46]对模型进行裁剪。
作为一个必然的结果,用户必须记住,basecaller在他们的数据集上的有效准确性可能比广告的准确性要低。
例如,ONT的基酶受体目前是在人类、酵母和细菌DNA的混合物上进行训练的;
它们在非cg甲基化丰富的植物DNA上的表现可能是[50]较低。
正如Guppy basecaller的定期更新所证明的那样, basecalling(碱基判定)仍是一个活跃的发展领域。
Errors, correction, and polishing
Both SMRT and nanopore technologies provide lower per read accuracy than short-read sequencing. In the case of SMRT, the circular consensus sequence quality is heavily dependent on the number of times the fragment is read—the depth of sequencing of the individual SMRTbell molecule (Fig. 1c)—a function of the length of the original fragment and longevity of the polymerase. With the Sequel v2 chemistry introduced in 2017, fragments longer than 10 kbp were typically only read once and had a single-pass accuracy of 85–87% [51]. The late 2018 v3 chemistry increases the longevity of the polymerase (from 20 to 30 kb for long fragments). An estimated four passes are required to provide a CCS with Q20 (99% accuracy) and nine passes for Q30 (99.9% accuracy) [18]. If the errors were non-random, increasing the sequencing depth would not be sufficient to remove them. However, the randomness of sequencing errors in subreads, consisting of more indels than mismatches [52–54], suggests that consensus approaches can be used so that the final outputs (e.g. CCS, assembly, variant calls) should be free of systematic biases. Still, CCS reads retain errors and exhibit a bias for indels in homopolymers [18].
On the other hand, the quality of nanopore reads is independent of the length of the DNA fragment. Read quality depends on achieving optimal translocation speed (the rate of ratcheting base by base) of the nucleic acid through the pore, which typically decreases in the late stages of sequencing runs, negatively affecting the quality [55]. Contrary to SMRT sequencing, a nanopore sequencing library is made of linear fragments that are read only once. In the most common, 1D sequencing protocol, each strand of the dsDNA fragment is read independently, and this single-pass accuracy is the final accuracy for the fragment. By contrast, the 1D2 protocol is designed to sequence the complementary strand in immediate succession of up to 75% of fragments, which allows the calculation of a more accurate consensus sequence for the library insert. To date, the median single-pass accuracy of 1D sequencing across a run can reach 95% (manufacturer’s numbers [56]). Release 6 of the human genomic DNA NA12878 reference data set reports 91% median accuracy [17]. 1D2 sequencing can achieve a median consensus accuracy of 98% [56]. An accurate consensus can also be derived from linear fragments if the same sequence is present multiple times: the concept of circularisation followed by rolling circle amplification for generating nanopore libraries is similar to SMRT sequencing, and subreads can be used to determine a high-quality consensus [57–59]. ONT is developing a similar linear consensus sequencing strategy based on isothermal polymerisation rather than circularisation [56].
Indels and substitutions are frequent in nanopore data, partly randomly but not uniformly distributed. Low-complexity stretches are difficult to resolve with the current (R9) pores and basecallers [56], as are homopolymer sequences. Measured current is a function of the particular k-mer residing in the pore, and because translocation of homopolymers does not change the sequence of nucleotides within the pore, it results in a constant signal that makes determining homopolymer length difficult. A new generation of pores (R10) was designed to increase the accuracy over homopolymers [56]. Certain k-mers may differ in how distinct a signal they produce, which can also be a source of systematic bias. Sequence quality is of course intimately linked to the basecaller used and the data that has been used to train it. Read accuracy can be improved by training the basecaller on data that is similar to the sample of interest [46]. ONT regularly release chemistry and software updates that improve read quality: 4 pore versions were introduced in the last 3 years (R9.4, R9.4.1, R9.5.1, R10.0), and in 2019 alone, there were 12 Guppy releases. PacBio similarly updates hardware, chemistry, and software: the last 3 years have seen the release of 1 instrument (Sequel II), 4 chemistries (Sequel v2 and v3; Sequel II v1 and v2), and 4 versions of the SMRT-LINK analysis suite.
Although current long-read accuracy is generally sufficient to uniquely determine the genomic origin of the read, certain applications require high base-level accuracy, including de novo assembly, variant calling, or defining intron-exon boundaries [54]. Two groups of methods to error correct long-reads can be employed: methods that only use long reads (non-hybrid) and methods that leverage the accuracy of additional short-read data (hybrid) (Fig. 2). Zhang et al. recently reviewed and benchmarked 15 of these long-read error correction methods [60], while Fu et al. focused on 10 hybrid error correction tools [61]. Lima et al. benchmarked 11 error correction tools specifically for nanopore cDNA reads [62].
错误、修正和润色
与短读测序相比,SMRT和纳米孔技术的每读精度都较低。
在SMRT中,循环一致序列的质量在很大程度上取决于片段被读取的次数——单个SMRTbell分子测序的深度(图1c)——原始片段的长度和聚合酶的寿命的函数。
随着2017年Sequel v2化学技术的引入,长度超过10 kbp的片段通常只能读取一次,单次扫描的准确度为85-87%[51]。
2018年底的v3化学反应延长了聚合酶的寿命(对于长片段,从20 kb延长到30 kb)。
估计需要4个通道来提供Q20(99%精度)的CCS和9个通道提供Q30(99.9%精度)[18]的CCS。
如果错误是非随机的,增加测序深度将不足以去除它们。
然而,子读序列中序列错误的随机性,包括更多的插入而不是不匹配[52-54],这表明可以采用一致的方法,从而使最终输出(例如CCS、组装、变体调用)不存在系统性偏差。
尽管如此,CCS读取仍然保留错误,并对同聚物[18]的indel显示出偏见。
另一方面,纳米孔的质量与DNA片段的长度无关。
读取质量取决于核酸通过孔的最佳易位速度(碱基对碱基的棘轮速率),通常在测序运行的后期会降低,对[55]的质量产生负面影响。
与SMRT测序相反,纳米孔测序库由线性片段组成,只能读取一次。
在最常见的1D测序协议中,dsDNA片段的每条链都是独立读取的,这种单次测序的准确性就是该片段最终的准确性。
相比之下,1D2协议的设计是直接连续排列互补链的75%的片段,这允许计算更准确的库插入的一致序列。
到目前为止,单次测序的单道精度中值可达95%(制造商编号[56])。
释放6人基因组DNA NA12878参考数据集报告91%的准确性[17]。
1D2测序的中位一致准确率为98%[56]。
如果同一序列多次出现,也可以从线性片段中获得准确的一致性:循环后滚动环扩增产生纳米孔文库的概念与SMRT测序相似,子序列可用于确定高质量的一致性[57-59]。
ONT正在开发一种类似的基于等温聚合而不是循环[56]的线性一致测序策略。
纳米孔数据中经常出现凹痕和取代现象,部分是随机的,但不是均匀分布的。
低延伸很难解决与当前(R9机型)毛孔和basecallers[56],均聚物序列。
测量电流的函数的特定k-mer驻留在毛孔,而且因为易位的均聚物不会改变孔隙内的核苷酸序列,它导致一个常数使得确定长度均聚物困难的信号。
设计了新一代孔(R10)以提高均聚物[56]的准确性。
某些k-mers可能在它们产生的信号的不同程度上存在差异,这也可能是系统性偏差的来源。
序列质量当然与所使用的基本工具和用于训练序列的数据密切相关。
通过在与感兴趣的样本[46]相似的数据上训练basecaller,可以提高读取精度。
ONT定期发布化学和软件更新来提高阅读质量:在过去的3年里推出了4个pore版本(R9.4, R9.4.1, R9.5.1, R10.0),仅在2019年,就有12个Guppy版本。
PacBio同样对硬件、化学和软件进行更新:在过去的3年里,我们发布了1个仪器(Sequel II)、4个化学(Sequel v2和v3;
续作II v1和v2),以及4个版本的SMRT-LINK分析套件。
虽然目前读的准确性一般足以唯一确定的基因组起源看,某些应用程序需要基准面精度高,包括新创组装、变体的召唤,或定义intron-exon边界[54]。
两组方法可以使用纠错long-reads:方法只使用长读(标价)和方法,利用附加短内容数据的准确性(混合)(图2)。张等人最近回顾和基准测试15这些读误差修正方法[60],而福等人关注10混合纠错工具[61]。
Lima等人对11种专门用于纳米孔cDNA读取的纠错工具进行了基准测试[62]。
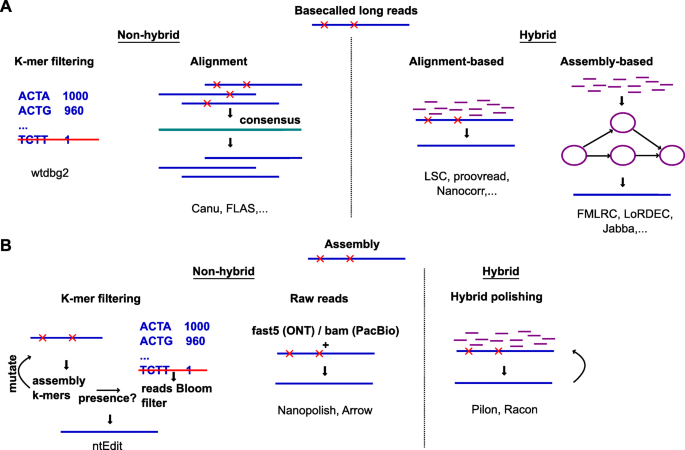
Paradigms of error correction (a) and polishing (b).
Errors in long reads and assembly are denoted by red crosses.
Non-hybrid methods only require long reads, while hybrid methods additionally require accurate short reads (purple)
错误校正(a)和抛光(b)的范例。
长read和组装中的错误用红色叉表示。
非混合方法只需要长读取,而混合方法额外需要精确的短读取(紫色)
In non-hybrid methods, all reads are first aligned to each other and a consensus is used to correct individual reads (Fig. 2a). These corrected reads can then be taken forward for assembly or other applications. Alternatively, because genomes only contain a small subset of all possible k-mers, rare k-mers in a noisy long-read data set are likely to represent sequencing errors. Filtering out these rare k-mers, as the wtdbg2 assembler does [63], effectively prevents errors from being introduced in the assembly (Fig. 2a).
Hybrid error correction methods can be further classified according to how the short reads are used. In alignment-based methods, the short reads are directly aligned to the long reads, to generate corrected long reads (Fig. 2a). In assembly-based methods, the short reads are first used to build a de Bruijn graph or assembly. Long reads are then corrected by aligning to the assembly or by traversing the de Bruijn graph (Fig. 2a). Assembly-based methods tend to outperform alignment-based methods in correction quality and speed, and FMLRC [64] was found to perform best in the two benchmark studies [60, 61].
After assembly, the process of removing remaining errors from contigs (rather than raw reads) is called ‘polishing’. One strategy is to use SMRT subreads through Arrow [65] or nanopore current traces through Nanopolish [66], to improve the accuracy of the consensus (Fig. 2b). For nanopore data, polishing while also taking into account the base modifications (as implemented for instance in Nanopolish [66]) further improves the accuracy of an assembly [46]. Alternatively, polishing can be done with the help of short reads using Pilon [67], Racon [68], or others, often in multiple rounds [50, 69, 70] (Fig. 2b). The rationale for iterative hybrid polishing is that as errors are corrected, previously ambiguously mapped short reads can be mapped more accurately. While certain pipelines repeat polishing until convergence (or oscillatory behaviour, where the same positions are changed back and forth between each round), too many iterations can decrease the quality of the assembly, as measured by the BUSCO score [71]. To increase scalability, ntEdit foregoes alignment in favour of comparing the draft assembly’s k-mers to a thresholded Bloom filter built from the sequencing reads [72] (Fig. 2b).
Despite continuous improvements in the accuracy of long reads, error correction remains indispensable in many applications. We identified 62 tools that are able to carry out error correction. There is no silver bullet, and correcting an assembly requires patience and careful work, often combining multiple tools (e.g. Racon, Pilon, and Nanopolish [50]). Adding to the difficulty of the absence of an authoritative error correction pipeline, certain tools do not scale well for deep sequencing or large genomes [50]. Furthermore, most tools are designed with haploid assemblies in mind. Allelic variation, repeats, or gene families may not be correctly handled.
在非混合方法中,首先将所有reads彼此对齐,并使用一致性来纠正单个reads(图2a)。
这些校正后的读数然后可以继续进行组装或其他应用。
另外,由于基因组只包含所有可能的k-mers的一个小子集,在长时间嘈杂的数据集中罕见的k-mers可能代表测序错误。
正如wtdbg2汇编程序所做的那样,过滤掉这些罕见的k-mers[63],可以有效地防止装配过程中引入错误(图2a)。
混合纠错方法可以根据短读的使用方式进一步分类。
在基于比对的方法中,短读直接与长读对齐,以生成校正后的长读(图2a)。
在基于集合的方法中,短读首先用于构建de Bruijn图或集合。
然后通过对准组件或遍历de Bruijn图(图2a)来校正长读数。
基于集合的方法在校正质量和速度上往往优于基于对准的方法,在两个基准研究中发现FMLRC[64]表现最好[60,61]。
组装后,从contigs(而不是原始读取)中删除剩余错误的过程称为“抛光”。
一种策略是通过箭头[65]使用SMRT子片段,或通过纳米微粒[66]使用纳米孔电流线,以提高一致性的准确性(图2b)。
对于纳米孔数据,抛光同时也考虑到碱基修改(例如在Nanopolish中实施[66])进一步提高组装[46]的精度。
另外,抛光也可以在短读的帮助下完成,如使用Pilon[67]、Racon[68]或其他,通常是多轮[50,69,70](图2b)。
迭代混合抛光的基本原理是,当错误被纠正后,以前模糊映射的短读可以被更准确地映射。
当某些管道重复抛光直到收敛(或振荡行为,相同的位置在每轮之间来回改变),太多的迭代会降低装配的质量,由BUSCO评分来衡量[71]。
为了提高可扩展性,ntEdit放弃了对k-mers的比对,而将草图的k-mers与从测序reads构建的阈值布隆过滤器进行比较[72](图2b)。
尽管长读的准确度不断提高,但在许多应用中,纠错仍然是必不可少的。
我们确定了62种能够进行纠错的工具。
没有什么银弹,而且纠正组装需要耐心和细致的工作,通常需要结合多种工具(如Racon, Pilon和Nanopolish[50])。
由于缺乏权威的纠错管道,某些工具在深度测序或大基因组[50]中不能很好地发挥作用,这增加了难度。
此外,大多数工具在设计时都考虑到单倍体组件。
等位基因变异、重复或基因家族可能不能正确处理。
Detecting structural variation
While short reads perform well for the identification of single nucleotide variants (SNVs) and small insertion and deletions (indels), they are not well suited to the detection of larger sequence changes [73]. Collectively referred to as structural variants (SVs), insertions, deletions, duplications, inversions, or translocations that affect ≥ 50 bp [74] are more amenable to long-read sequencing [75, 76] (Fig 1c). Because of these past technical limitations, structural variants have historically been under-studied despite being an important source of diversity between genomes and relevant for human health [77, 78].
The ability of long reads to span repeated elements or repetitive regions provides unique anchors that facilitate de novo assembly and SV calling [73]. Even relatively short (5 kb) SMRT reads can identify structural variants in the human genome that were previously missed by short-read technologies [79]. Obtaining deep coverage of mammalian-sized genomes with long reads remains costly; however, modest coverage may be sufficient: 8.6 × SMRT sequencing [14] and 15–17 × nanopore sequencing [80, 81] have been shown to be effective in detecting pathogenic variants in humans. Heterozygosity or mosaicism naturally increase the coverage requirements.
Evaluating the performance of long-read SV callers is complicated by the fact that benchmark data sets may be missing SVs in their annotation [73, 77], especially when it comes only from short reads. Therefore, validation of new variants has to be performed via other methods. Developing robust benchmarks is an ongoing effort [82], as is devising solutions to visualise complex, phased variants for critical assessment [82, 83].
For further details on structural variant calling from long-read data, we refer the reader to two recent reviews: Mahmoud et al. [73] and Ho et al. [77].
检测结构变化
虽然短读段可以很好地识别单核苷酸变异(SNVs)和小插入和删除(indels),但它们不太适合检测更大的序列变化[73]。
被统称为结构变异(SVs)的插入、缺失、重复、逆序或易位影响≥50 bp[74]更适合长读测序[75,76](图1c)。
由于这些过去的技术限制,尽管结构变异是基因组之间多样性的重要来源,并且与人类健康相关,但它一直没有得到足够的研究[77,78]。
长读取跨越重复元素或重复区域的能力提供了唯一的锚点,便于从头组装和SV调用[73]。
即使是相对较短的(5 kb) SMRT读取,也可以识别出之前被短读取技术忽略的人类基因组中的结构变异[79]。
要获得哺乳动物大小的基因组的长读码深度覆盖仍然代价高昂;
然而,适度的覆盖可能就足够了:8.6倍SMRT测序[14]和15-17倍纳米孔测序[80,81]已被证明在检测人类致病变异中有效。
杂合性或马赛克 自然增加了覆盖要求。
由于基准数据集的注释中可能缺少SV,评估长读取SV调用者的性能变得复杂[73,77],特别是当它仅来自短read时。
因此,新变体的验证必须通过其他方法来执行。
开发健壮的基准是一个持续的努力[82],想象复杂的设计解决方案,分阶段关键评估变异(82、83)。
关于长读数据的结构变体调用的更多细节,我们请读者参阅最近的两篇综述:Mahmoudet al.[73]和Hoet al.[77]。
Detecting base modifications
In addition to the canonical A, T, C, and G bases, DNA can contain modified bases that vary in nature and frequency across organisms and tissues. N-6-methyladenine (6mA), 4-methylcytosine (4mC), and 5-methylcytosine (5mC) are frequent in bacteria. 5mC is the most common base modification in eukaryotes, while its oxidised derivatives 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxycytosine (5caC) are detected in certain mammalian cell types but have yet to be deeply characterised [84–88]. Still, more base modifications that result from DNA damage occur at a low frequency [87].
The nucleotides that compose RNA are even more varied. Over 150 modified bases have been documented to date [89, 90]. These modifications also have functional roles, for example, in mRNA stability [91], transcriptional repression [92], and translational efficiency [93]. However, most RNA modifications remain ill-characterised due to technological limitations [94]. Aside from the modifications to standard bases, base analogues may also be introduced to nucleic acids, such as the thymidine analogue BrdU which is used to track genomic replication [95].
Mapping of nucleic acid modifications has traditionally relied on specific chemical treatment (e.g. bisulfite conversion that changes unmethylated cytosines to uracils [96]) or immunoprecipitation followed by sequencing [97]. The ability of the long-read platforms to sequence native nucleic acids provides the opportunity to determine the presence of many more modifications, at base resolution in single molecules, and without specialised chemistries that can be damaging to the DNA [98]. Long reads thus allow the phasing of base modifications along individual nucleic acids, as well as their phasing with genetic variants, opening up opportunities in exploring epigenetic heterogeneity [34, 99]. Long reads also enable the analysis of base modifications in repetitive regions of the genome (centromeres or transposons), where short reads cannot be mapped uniquely.
In SMRT sequencing, base modifications in DNA or RNA [100, 101] are inferred from the delay between fluorescence pulses, referred to as interpulse duration (IPD) [98] (Fig. 3). Base modifications impact the speed at which the polymerase progresses, at the site of modification and/or downstream. Comparison with the signal from an in silico or non-modified reference (e.g. amplified DNA) suggests the presence of modified bases [102, 103]. It is notably possible to detect 6mA, 4mC, 5mC, and 5hmC DNA modifications, although at different sensitivity. Reliable calling of 6mA and 4mC requires 25 × coverage per strand, whereas 250 × is required for 5mC and 5hmC, which have subtler impacts on polymerase kinetics [102]. Such high coverage is not realistic for large genomes and does not allow single-molecule epigenetic analysis. Coverage requirements can be reduced by conjugating a glucose moeity to 5hmC, which gives a stronger IPD signal during SMRT sequencing [102, 103]. Polymerase dynamics and base modifications can be analysed directly via the SMRT Portal, or for more advanced analyses with R-kinetics, kineticsTools or basemods [104]. SMALR [99] is dedicated to the detection of base modifications in single SMRT reads.
检测基础上修改
除了典型的A、T、C和G碱基,DNA还可以包含在不同生物和组织中性质和频率不同的修饰碱基。
n -6-甲基ladenine (6mA)、4-甲基胞嘧啶(4mC)和5-甲基胞嘧啶(5mC)在细菌中常见。
5 mc是最常见的基础修改在真核生物中,而其氧化衍生物5-hydroxymethylcytosine (5 hmc), 5-formylcytosine (5 fc),和5-carboxycytosine (5 cac)检测在某些哺乳动物细胞类型但尚未深入特征[84 - 88]。
然而,更多的碱基修饰是由DNA损伤导致的,其发生频率较低[87]。
组成RNA的核苷酸甚至更多样化。
到目前为止,已有超过150个修改的碱基被记录在案[89,90]。
这些修饰也有功能作用,例如,在mRNA的稳定性[91]、转录抑制[92]和翻译效率[93]。
然而,由于技术上的限制,大多数RNA修饰的特征仍然很差[94]。
除了对标准碱基的修饰,碱基类似物也可能被引入核酸中,如用于跟踪基因组复制的胸苷类似物BrdU[95]。
核酸修饰的定位传统上依赖于特定的化学处理(例如亚硫酸氢盐转化将未甲基化的胞嘧啶转化为尿嘧啶[96])或免疫沉淀后测序[97]。
长时间阅读的平台能够对天然核酸进行排序,这为确定更多修饰的存在提供了机会,在单分子的碱基分辨率下,并且没有可能损害DNA的特殊化学物质[98]。
因此,长读本允许碱基修饰随单个核酸的分期,以及它们与遗传变异的分期,为探索表观遗传异质性提供了机会[34,99]。
长读子还能分析基因组重复区域(着丝粒或转座子)的碱基修饰,而短读子不能被唯一地映射。
在SMRT测序中,DNA或RNA中的碱基修饰[100,101]是从荧光脉冲之间的延迟(简称脉冲间持续时间(IPD))中推断出来的[98](图3)。碱基修饰影响聚合酶在修饰位点和/或下游的进展速度。
与来自含硅或未修饰参考物(如扩增的DNA)的信号比较表明,修饰碱基的存在[102,103]。
虽然在不同的灵敏度下,检测6mA、4mC、5mC和5hmC的DNA修饰是明显可能的。
6mA和4mC的可靠召唤需要每链25倍的覆盖度,而5mC和5hmC需要250倍的覆盖度,这对聚合酶动力学有更微妙的影响[102]。
如此高的覆盖率对于大的基因组是不现实的,也不允许单分子表观遗传分析。
通过将葡萄糖摩尔与5hmC结合,可以降低覆盖度要求,这在SMRT测序过程中提供了更强的IPD信号[102,103]。
聚合酶动力学和碱基修饰可以通过SMRT门户直接分析,也可以通过r -动力学、动力学粪便或碱基进行更高级的分析[104]。
SMALR[99]致力于检测单个SMRT读取中的碱基修改。
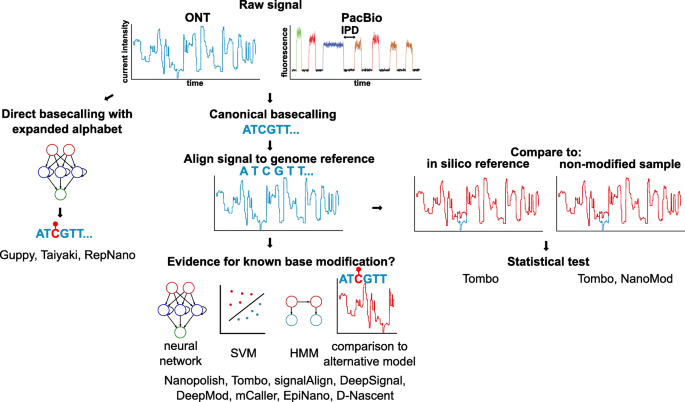
Methods to detect base modifications in long-read sequencing.
Base modifications can be inferred from their effect on the current intensity (nanopore) and inter-pulse duration (IPD, SMRT).
Strategies to call base modifications in nanopore sequencing and the corresponding tools are further depicted
方法检测长读测序中的碱基修饰。
碱基修饰可以从它们对电流强度(纳米孔)和脉冲间持续时间(IPD, SMRT)的影响中推断出来。
在纳米孔测序中调用碱基修饰的策略和相应的工具进一步描述
In nanopore sequencing, modified RNA or DNA bases affect the flow of the current through the pore differently than non-modified bases, resulting in signal shifts (Fig. 3). These shifts can be identified post-basecalling and post-alignment with three distinct methods: (a) without prior knowledge about the modification (de novo) by comparing to an in silico reference [105], or a control, non-modified sample (typically amplified DNA) [105, 106]; (b) using a pre-trained model [66, 107, 108] (Fig. 3, Table 1); and (c) directly by a basecaller using an extended alphabet [45, 109].
在纳米孔测序中,修饰过的RNA或DNA碱基与未修饰过的碱基对孔内电流的影响不同,从而导致信号偏移(图3)。这些偏移可以通过三种不同的方法来识别:
(a)在事先不知道与in silicon参考文献[105]相比的修改(de novo)的情况下,
或对照、非修饰样本(通常是扩增的DNA) [105,106];
(b)使用预训练模型[66,107,108](图3,表1);
和(c)直接由basic basic aller使用扩展的字母表[45,109]。
表1纳米孔数据基修改检测工具和策略
(HMM hidden Markov model, HPD hierarchical Dirichlet process, CNN convolutional neural network,
LSTM long short-term memory, RNN recurrent neural network, SVM support vector machine)
De novo approaches, as implemented by Tombo [105] or NanoMod [106], allow the discovery of modifications and modified motifs by statistically testing the deviation of the observed signal relative to a reference. However these methods suffer from a high false discovery rate and are not reliable at the single-molecule level. The comparison to a control sample rather than an in silico reference increases the accuracy of detection, but requires the sequencing of twice as many sample as well as high coverage to ensure that genomic segments are covered by both control and test sample reads. De novo calling of base modifications is limited to highlighting regions of the genomes that may contain modified bases, without being able to reveal the precise base or the nature of the modification.
Pre-trained models interrogate specific sites and classify the data as supporting a modified or unmodified base. Nanopolish [66] detects 5mC with a hidden Markov model, which in signalAlign [107] is combined with a hierarchical Dirichlet process, to determine the most likely k-mer (modified or unmodified). D-NAscent [95] utilises an approach similar to Nanopolish to detect BrdU incorporation, while EpiNano uses support vector machines (SVMs) to detect RNA m6A. Recent methods use neural network classifiers to detect 6mA and 5mC (mCaller [108], DeepSignal [110], DeepMod [111]). The accuracy of these methods is upwards of 80% but varies between modifications and motifs. Appropriate training data is crucial and currently a limiting factor. Models trained exclusively on samples with fully methylated or unmethylated CpGs will not perform optimally on biological samples with a mixture of CpG and mCpGs, or 5mC in other sequence contexts [66, 105]. Low specificity is particularly problematic for low abundance marks. m6A is present at 0.05% in mRNA [113, 114]; therefore, a method testing all adenosines in the transcriptome with sensitivity and specificity of 90% at the single-molecule, single-base level would result in an unacceptable false discovery rate of 98%.
Direct basecalling of modified bases is a recent addition to ONT’s basecaller Guppy, currently limited to 5mC in the CpG context. A development basecaller, Taiyaki [45], can be trained for specific organisms or base modifications. RepNano can basecall BrdU in addition to the four canonical DNA bases [109]. Two major bottlenecks in the creation of modification-ready basecallers are the need for appropriate training data and the combinatorial complexity of adding bases to the basecalling alphabet. There is also a lack of tools for the downstream analysis of base modifications: most tools output a probability that a certain base is modified, while traditional differential methylation algorithms expect binary counts of methylated and unmethylated bases.
从头方法,如Tombo[105]或NanoMod[106]所实现的,允许通过统计测试观察到的信号相对于参考的偏差来发现修改和修改的基模。
但这些方法存在误发现率高,在单分子水平上不可靠的问题。
与对照样本而非in硅片对照的比较提高了检测的准确性,但需要两倍于对照样本的测序量和高覆盖率,以确保对照样本和检测样本都能覆盖基因组片段。
碱基修饰的重新调用仅限于突出可能包含修饰碱基的基因组区域,而不能揭示精确的碱基或修饰的性质。
预先训练过的模型查询特定的站点,并将数据分类为支持已修改或未修改的数据库。
Nanopolish[66]使用隐藏马尔科夫模型检测5mC,在signalAlign[107]中,该模型与分层的Dirichlet过程相结合,以确定最有可能的k-mer(修改或未修改)。
D-NAscent [95]使用类似于Nanopolish的方法检测BrdU的加入,而EpiNano使用支持向量机(SVMs)检测RNA m6A。
最近的方法使用神经网络分类器检测6mA和5mC (mCaller [108], DeepSignal [110], DeepMod[111])。
这些方法的精确度高达80%,但因修改和图案的不同而有所不同。
适当的培训数据是至关重要的,也是目前的一个限制因素。
仅针对完全甲基化或未甲基化CpGs的样本进行训练的模型,在CpG和mcpg混合的生物样本,或在其他序列背景下5mC的生物样本上的表现都不最佳[66,105]。
低特异性对于低丰度标记尤其成问题。
m6A在mRNA中以0.05%的比例存在[113,114];
因此,在单分子、单碱基水平上检测转录组中所有腺苷的敏感性和特异性均为90%的方法,其错误发现率高达98%,难以接受。
直接碱基化修改的碱基是最近在ONT的basecaller Guppy中添加的,目前在CpG环境中限制为5mC。
Taiyaki[45]是一种发育型碱基诱变剂,可以被训练用于特定的生物体或碱基修饰。
除了四个典型的DNA碱基外,RepNano还能碱基BrdU[109]。
在创建修改准备基库的两个主要瓶颈是需要适当的训练数据和组合复杂性添加基到基库字母表。
也缺乏对碱基修改进行下游分析的工具:
大多数工具输出一个特定碱基被修改的概率,而传统的差异甲基化算法期望甲基化和非甲基化碱基的二进制计数。
Analysing long-read transcriptomes
Alternative splicing is a major mechanism increasing the complexity of gene expression in eukaryotes [115, 116]. Practically, all multi-exon genes in humans are alternatively spliced [117, 118], with variations between tissues and between individuals [119]. However, fragmented short reads cannot fully assemble nor accurately quantify the expressed isoforms, especially at complex loci [120, 121]. Long-read sequencing provides a solution by ideally sequencing full-length transcripts. Recent studies that used bulk, single-cell, or targeted long-read sequencing suggest that our best transcript annotations are still missing vast numbers of relevant isoforms [122–126]. As noted above, sequencing native RNA further provides the opportunity to better characterise RNA modifications or other characteristics such as poly-A tail length. Despite its many promises, analysis of long-read transcriptomes remains challenging. Few of the existing tools for short-read RNA-seq analysis are able to appropriately deal with the high error rate of long reads, necessitating the development of dedicated tools and extensive benchmarks. Although recently, the field of long-read transcriptomics is rapidly expanding, we tallied 36 tools related to long-read transcriptome analysis (Fig. 1b).
Most long-read isoform detection tools work by clustering aligned and error-corrected reads into groups and collapsing these into isoforms, but the detailed implementations differ between tools (Fig. 4). PacBio’s Iso-Seq3 [127, 128] is the most mature pipeline for long-read transcriptome analysis, allowing the assembly of full-length transcripts. It performs pre-processing for SMRT reads, de novo discovery of isoforms by hierarchical clustering and iterative merging, and polishing. Cupcake [129] provides scripts for downstream analysis such as collapsing redundant isoforms and merging Iso-Seq runs from different batches, giving abundance information as well as performing junction analysis. In the absence of a reference genome, Iso-Seq can assemble a transcriptome, but transcripts from related genes may be merged [130] as a trade-off for correcting reads with a high error rate. Furthermore, the library preparation for Iso-Seq usually requires size fractionation, which makes absolute and relative quantification difficult. The per-read cost remains high, making well-replicated differential expression study designs prohibitively expensive.
分析读转录组
可变剪接是增加真核生物基因表达复杂性的主要机制[115,116]。
实际上,人类所有的多外显子基因都是交替剪接的[117,118],在组织和个体之间存在差异[119]。
然而,片段短读不能完全组装或准确量化表达的异构体,特别是在复杂位点[120,121]。
长读测序提供了一个解决方案,理想的测序全长转录。
最近使用大容量、单细胞或靶向长读测序的研究表明,我们最好的转录子注释仍然缺少大量相关亚型[122-126]。
如上所述,对原生RNA进行测序进一步提供了更好地表征RNA修饰或其他特征(如多a尾长度)的机会。
尽管有许多承诺,分析长期阅读的转录本仍然具有挑战性。
现有的用于短读RNA-seq分析的工具很少能够适当地处理长读的高错误率,因此需要开发专用工具和广泛的基准测试。
虽然最近长读转录组学领域正在迅速扩大,但我们统计了36种与长读转录组分析相关的工具(图1b)。
大多数读同种型检测工具通过集群对齐和error-corrected read到团体和崩溃这些亚型,但是详细的实现不同工具之间(图4),PacBio Iso-Seq3(127、128)是最成熟的管道读转录组分析,允许装配完整的记录。
它执行SMRT读取的预处理,通过分层聚类和迭代合并重新发现亚型,以及抛光。
Cupcake[129]提供了用于下游分析的脚本,例如折叠冗余的异型和合并来自不同批次的Iso-Seq运行,提供了丰富的信息并执行连接分析。
在没有参考基因组的情况下,Iso-Seq可以组装一个转录组,但可能会合并相关基因的转录本[130],以弥补较高错误率的误读。
此外,异构seq的库准备通常需要对大小进行分馏,这使得绝对定量和相对定量都很困难。
每次读取的成本仍然很高,使得复制良好的差异表达研究设计昂贵得令人望而却步。
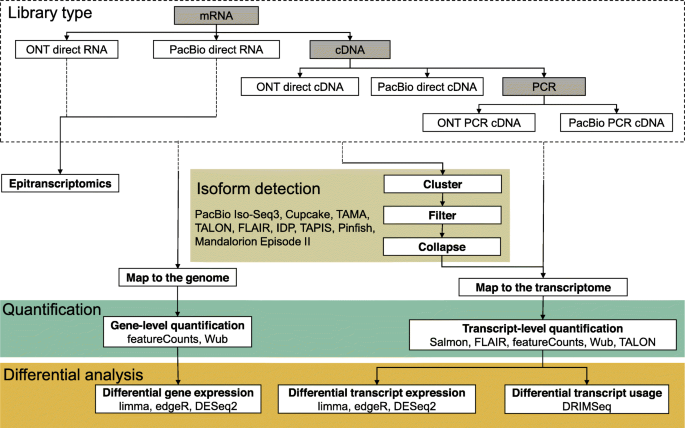
Types of transcriptomic analyses and their steps. The choice of sequencing protocol amongst the six available workflows affects the type, characteristics, and quantity of data generated. Only direct RNA sequencing allows epitranscriptomic studies, but SMRT direct RNA sequencing is a custom technique that is not fully supported. The remaining non-exclusive applications are isoform detection, quantification, and differential analysis. The dashed lines in arrows represent upstream processes to transcriptomics
转录分析的类型及其步骤。
在六个可用工作流中选择排序协议会影响生成的数据的类型、特征和数量。
只有直接RNA测序可以进行表转录组研究,但SMRT直接RNA测序是一种定制技术,不完全支持。
其余的非专用应用是同位素检测、定量和鉴别分析。
箭头中的虚线表示转录组的上游过程
Alternative isoform detection pipelines such as IsoCon [130], SQANTI [131], and TALON [132] attempt to mitigate the erroneous merging of similar transcripts of the Iso-Seq pipeline. IsoCon and SQANTI specifically work with SMRT data while TALON is a technology-independent approach. IsoCon uses the full-length transcripts from Iso-Seq to perform clustering and partial error correction and identify candidate transcripts without losing potential true variants within each cluster. SQANTI generates quality control reports for SMRT Iso-Seq data and detects and removes potential artefacts. TALON, on the other hand, relies heavily on the GENCODE annotation. Since both IsoCon and TALON focus on the human genome, they may not perform equally well with genomes from non-model organisms. A number of alternative isoform annotation pipelines for SMRT and/or nanopore data have recently emerged, such as FLAIR [133], Tama [134], IDP [122], TAPIS [135], Mandalorion Episode II [36, 57], and Pinfish [136]. Some of them use short reads to improve exon junction annotation. However, their accuracy has not yet been extensively tested.
In addition to high error rates, potential coverage biases are currently not explicitly taken into account by long-read transcriptomic tools. In ONT’s direct RNA sequencing protocol, transcripts are sequenced from the 3′ to the 5′ end; therefore, any fragmentation during the library prep, or pore blocking, results in truncated reads. In our experience, it is common to see a coverage bias towards the 3′ end of transcripts, which can affect isoform characterisation and quantification. Methods that sequence cDNA will also show these coverage biases due to fragmentation and pore-blocking (for nanopore data), compounded by non-processivity of the reverse transcriptase [124], more likely to stall when it encounters RNA modifications [137]. Finally, the length-dependent or sequence-dependent biases introduced by protocols that rely on PCR are currently not well characterised nor accounted for.
To quantify the abundance of transcripts or genes, several methods can be used (Fig. 4). Salmon’s [138] quasi-mapping mode quantifies reads directly against a reference index, and its alignment-based mode instead works with aligned sequences. The Wub package [139] also provides a script for read counting. The featureCounts [140] function from the Subread package [141, 142] supports long-read gene level counting. The FLAIR [133] pipeline provides wrappers for quantifying FLAIR isoform usage across samples using minimap2 or Salmon. Of course, for accurate transcript-level quantification, these methods rely on a complete and accurate isoform annotation; this is currently the difficult step.
Two types of differential analyses can be run: gene level or transcript level (Fig. 4). Transcript-level analyses may be further focused on differential transcript usage (DTU), where the gene may overall be expressed at the same level between two conditions, but the relative proportions of isoforms may vary. The popular tools for short-read differential gene expression analysis, such as limma [143], edgeR [144, 145], and DESeq2 [146], can also be used for long-read differential isoform or gene expression analyses. DRIMSeq [147] can perform differential isoform usage analysis using the Dirichlet-multinomial model. One difference between short- and long-read counts is that for the latter, counts per million (cpm) are effectively transcripts per million (tpm), whereas for short reads (and random fragmentation protocols), transcript length influences the number of reads, and therefore, cpms need scaling by transcript length to obtain tpms. The biological interpretation of differential isoform expression strongly depends on the classification of the isoforms, for example, whether the isoforms code for the same or different proteins or whether premature stop codons make them subject to nonsense-mediated decay. This is currently not well integrated into the analyses.
替代的isoform检测管道,如IsoCon[130]、SQANTI[131]和TALON[132],试图减轻Iso-Seq管道相似转录本的错误合并。
IsoCon和SQANTI专门与SMRT数据工作,而TALON是一个技术独立的方法。
IsoCon使用来自Iso-Seq的全长转录本进行聚类和部分纠错,并在不丢失每个聚类内潜在的真实变异的情况下识别候选转录本。
SQANTI生成质量控制报告SMRT Iso-Seq数据和检测和消除潜在的文物。
另一方面,TALON在很大程度上依赖于GENCODE注释。
由于IsoCon和TALON都专注于人类基因组,他们可能在处理非模式生物的基因组时表现不太好。
最近出现了许多用于SMRT和/或纳米孔数据的替代异型注释管道,如FLAIR[133]、Tama[134]、IDP[122]、TAPIS[135]、Mandalorion Episode II[36,57]和Pinfish[136]。
其中一些使用短读来改进外显子结注释。
然而,它们的准确性还没有得到广泛的测试。
除了高错误率,潜在的覆盖偏差目前没有被长期阅读的转录组工具明确地考虑。
在ONT的直接RNA测序方案中,转录本从3’端到5’端进行测序;
因此,在库准备期间的任何片段,或孔阻塞,都会导致读取被截断。
根据我们的经验,通常看到报道偏向3的成绩单,可影响同种型描述和量化。
对cDNA进行测序的方法也会由于片段和孔阻塞(对于纳米孔数据)而出现覆盖偏差,再加上逆转录酶的非可加工性[124],当其遇到RNA修饰时更有可能出现停顿[137]。
最后,依赖于PCR的协议引入的依赖于长度或序列的偏差目前还没有很好地描述和解释。
为了量化转录本或基因的丰度,可以使用几种方法(图4)。Salmon[138]的准映射模式直接根据参考指数来量化reads,而基于比对的模式则与已比对的序列一起工作。
Wub包[139]还提供了一个剧本读计数。
Subread包[141,142]中的featucounts[140]功能支持长读基因水平计数。
FLAIR[133]管道提供了使用minimap2或Salmon跨样本定量FLAIR isoform使用的包装器。
当然,为了准确的转录水平量化,这些方法依赖于一个完整和准确的isoform注释;
这是目前困难的一步。
可以进行两种类型的差异分析:基因水平或转录水平(图4)。转录水平分析可以进一步集中于差异转录使用(DTU),其中基因在两种条件下可能总体表达在相同水平,但亚型的相对比例可能不同。
流行的工具短内容差异基因表达分析,如limma[143],磨边机(144、145),和DESeq2[146],也可以用来读微分同种型或基因表达分析。
DRIMSeq[147]可以使用dirichlet -多项模型进行微分异型使用分析。
短读和长读计数的一个区别是,对于后者,每百万的计数(cpm)实际上是每百万的转录本(tpm),而对于短读(和随机片段协议),转录本长度影响读取的数量,因此,cpm需要根据转录本长度进行缩放以获得tpms。
不同亚型表达的生物学解释在很大程度上取决于亚型的分类,例如,是否亚型编码相同或不同的蛋白质,或过早停止密码子是否使他们受到无意义的介导的衰变。
这一点目前没有很好地集成到分析中。
Combining long reads, synthetic long reads, and short reads
Assemblies based solely on long reads generally produce highly complete and contiguous genomes [148–150]; however, there are many situations where short reads or reads generated from synthetic long-read technology further improve the results [151–153].
Different technologies can intervene at different scales: short reads ensure base-level accuracy, high-quality 5–15-kb SMRT reads generate good contigs, while ultra-long (100 kb+) nanopore reads, optical mapping or Hi-C improve scaffolding of the contigs into chromosomes [11, 17, 154–157]. Combining all of these technologies in a single genomic project would be costly. Instead, combinations of subsets are frequent, in particular, nanopore/SMRT with short-read sequencing [50, 152, 153, 158], although other combinations can be useful. Nanopore assembly of wild strains of Drosophila melanogaster supported by scaffolds generated from Hi-C corrected two misalignments of contigs in the reference assembly [154]. Optical maps helped resolve misassembly of SMRT-based chromosome level contigs of three plant relatives of Arabidopsis thaliana, where unrelated parts of the genome were erroneously linked [155].
For structural variation or base modification detection, obtaining orthogonal support from SMRT and nanopore data is valuable to confirm discoveries and limit false positives [77, 108, 159]. The error profiles of SMRT and nanopore sequencing are not identical—though both technologies experience difficulty around homopolymers—combining them can draw on their respective strengths.
Certain tools such as Unicycler [160] integrate long- and short-read data to produce hybrid assemblies, while other tools have been presented as pipelines to achieve this purpose (e.g. Canu, Pilon, and Racon in the ont-assembly-polish pipeline [45]). Still, combining tools and data types remains a challenge, usually requiring intensive manual integration.
结合长读、综合长读和短读
单独基于长读的装配通常产生高度完整和连续的基因组[148-150];
然而,在很多情况下,短读或合成长读技术生成的读进一步提高了结果[151-153]。
不同的技术可以在不同的尺度上进行干预:短读取确保了基本水平的准确性,高质量的5- 15 kb SMRT读取产生良好的contigs,而超长(100 kb+)纳米孔读取、光学绘图或高分辨率扫描可以提高contigs进入染色体的支架[11,17,154 - 157]。
将所有这些技术结合在一个单一的基因组计划将是昂贵的。
相反,亚群的组合是常见的,特别是纳米孔/SMRT与短读测序[50,152,153,158],尽管其他组合可能有用。
由Hi-C生成的支架支撑的野生果蝇纳米孔组装纠正了文献组装中的两个contigs错位[154]。
光学地图帮助解决错误装配SMRT-based染色体水平重叠群的三个植物拟南芥的亲属,在基因组中不相关的部分错误链接[155]。
结构变化或基础修改检测,获得正交SMRT和纳米孔的支持数据是有价值的确认发现并限制假阳性(77、108、159)。
SMRT和纳米孔测序的误差并不相同——尽管这两种技术在均聚物方面都遇到了困难——将它们结合起来可以发挥各自的优势。
某些工具,如Unicycler[160],集成了长读和短读数据来生产混合组件,而其他工具则被作为管道来实现这一目的(例如,在ont-assembly-polish管道[45]中的Canu、Pilon和Racon)。
但是,组合工具和数据类型仍然是一个挑战,通常需要密集的手工集成。
long-read-tools.org: a catalogue of long-read sequencing data analysis tools
The growing interest in the potential of long reads in various areas of biology is reflected by the exponential development of tools over the last decade (Fig. 1a). There are open-source static catalogues (e.g. github.com/B-UMMI/long-read-catalog), custom pipelines developed by individual labs for specific purposes (e.g. Search results from GitHub), and others that attempt to generalise them for a wider research community [46]. Being able to easily identify what tools exist—or do not exist—is crucial to plan and perform best-practice analyses, build comprehensive benchmarks, and guide the development of new software.
For this purpose, we introduce long-read-tools.org, a timely database that comprehensively collates tools used for long-read data analysis. Users can interactively search tools categorised by technology and intended type of analysis. In addition to true long-read sequencing technologies (SMRT and nanopore), we include synthetic long-read strategies (10X linked reads, Hi-C, and Bionano optical mapping). The fast-paced evolution of long-read sequencing technologies and tools also means that certain tools become obsolete. We include them in our database for completeness but indicate when they have been superseded or are no longer maintained.
long-read-tools.org is an open-source project under the MIT License, whose code is available through GitHub [161]. We encourage researchers to contribute new database entries of relevant tools and improvements to the database, either directly via the GitHub repository or through the submission form on the database webpage.
long-read-tools.org:
长时间阅读的序列数据分析工具的目录
在过去的十年中,工具的指数级发展反映了人们对生物学各个领域的长阅读潜力的日益增长的兴趣(图1a)。
有开源的静态目录(例如github.com/B-UMMI/long-read-catalog),由个别实验室为特定目的(例如GitHub的搜索结果)开发的定制管道,以及其他一些试图将它们推广到更广泛的研究社区[46]的方法。
对于计划和执行最佳实践分析、构建全面的基准,以及指导新软件的开发,能够轻松地确定存在或不存在什么工具是至关重要的。
为此,我们引入了long-read-tools.org,这是一个及时的数据库,全面整理用于长时间阅读数据分析的工具。
用户可以交互搜索工具分类的技术和预期类型的分析。
除了真正的长读测序技术(SMRT和nanopore),我们还包括合成长读策略(10倍链读、Hi-C和Bionano光学绘图)。
测序技术和工具的快速发展也意味着某些工具已经过时。
为了完整性,我们将它们包括在我们的数据库中,但是当它们被取代或不再被维护时,我们会指出。
longread -tools.org是麻省理工学院许可的一个开源项目,其代码可通过GitHub[161]获得。
我们鼓励研究人员直接通过GitHub存储库或通过数据库网页上的提交表单,为数据库贡献相关工具和改进的新数据库条目。
Discussion
At the time of writing, for about USD1500, one can obtain around 30 Gbases of ≥ 99% accurate SMRT CCS (1 Sequel II 8M SMRT cell) or 50–150 Gbases of noisier but potentially longer nanopore reads (1 PromethION flow cell). While initially, long-read sequencing was perhaps most useful for assembly of small (bacterial) genomes, the recent increases in throughput and accuracy enable a broader range of applications. The actual biological polymers that carry genetic information can now be sequenced in their full length or at least in fragments of tens to hundreds of kilobases, giving us a more complete picture of genomes (e.g. telomere-to-telomere assemblies, structural variants, phased variations, epigenetics, metagenomics) and transcriptomes (e.g. isoform diversity and quantity, epitranscriptomics, polyadenylation).
These advances are underpinned by an expanding collection of tools that explicitly take into account the characteristics of long reads, in particular, their error rate, to efficiently and accurately perform tasks such as preprocessing, error correction, alignment, assembly, base modification detection, quantification, and species identification. We have collated these tools in the long-read-tools.org database.
The proliferation of long-read analysis tools revealed by our census makes a compelling case for complementary efforts in benchmarking. Essential to this process is the generation of publicly available benchmark data sets where the ground truth is known and whose characteristics are as close as possible to those of real biological data sets. Simulations, artificial nucleic acids such as synthetic transcripts or in vitro-methylated DNA, resequencing, and validation endeavours will all contribute to establishing a ground truth against which an array of tools can be benchmarked. In spite of the rapid iteration of technologies, chemistries, and data formats, these benchmarks will encourage the emergence of best practices.
A recurrent challenge in long-read data analysis is scalability. For instance in genome assembly, Canu [69] produces excellent assemblies for small genomes but takes too long to run for large genomes. Fast processing is crucial to enable parameter optimisation in applications that are not yet routine. The recently released wtdbg2 [63], TULIP [70], Shasta [162], Peregrine [163], Flye [164], and Ra [165] assemblers are orders of magnitude faster and are quickly being adopted. Similarly, for mapping long reads, minimap2’s speed, in addition to its accuracy, has contributed to its fast and wide adoption. Nanopolish [66] is popular both for assembly correction and base modification detection; however, it is slow on large data sets. The refactoring of its call-methylation function in f5c tool greatly facilitates work with large genomes or data sets [166].
Beyond data processing speed, scalability is also impacted by data generation, storage, and integration. Nanopore sequencing presents the fastest turnaround time. Once DNA is extracted, sequencing is underway in a matter of minutes to hours, and the PromethION sequencer provides adjustable high throughput with individually addressable parallel flow cells. All other library preparation procedures are more labour intensive, and sequencing may have to await pooling to fill a run, and flow cells need to be run in succession rather than in parallel. The raw nanopore data is however extremely voluminous (about 20 bytes per base), leading to substantial IT costs for large projects. SMRT movies are not saved for later re-basecalling, and the sequence and kinetic information takes up a smaller 3.5 bytes per base. Furthermore, hybrid methods incorporating strengths from other technologies such as optical mapping (Bionano, OpGen) and Hi-C add to the cost and analytical complexity of genomic projects. For these, manual data integration is a significant bottleneck, but the rewards are worth the effort.
Despite increasing accuracy of both SMRT and nanopore sequencing platforms, error correction remains an important step in long-read analysis pipelines. Published assemblies that omit careful error correction are likely to predict many spurious truncated proteins [167]. Hybrid error correction, leveraging the accuracy of short reads, is still outperforming long-read-only correction [60]. Modern short-read sequencing protocols require small input amounts (some even scale down to single cells) so sample amount is usually not a barrier to combining short- and long-read sequencing. Removing the need for short reads, and higher coverage via improvements in non-hybrid error correction tools and/or long-read sequencing accuracy, would reduce the cost, length, and complexity of genomic projects.
The much anticipated advances in epigenetics/epitranscriptomics promised by long-read sequencing are still in development. Many modifications, including 5mC, do not influence the SMRT polymerase’ dynamics sufficiently to be detected at a useful sensitivity (5mC requires 250 × coverage). In this case, software improvements are unlikely to yield significant gains, and improvements in sequencing chemistries are probably required [168]. Nanopore sequencing appears more amenable to the detection of a wide array of base modifications (to date: 5mCG, BrdU, 6mA), but the lack of ground truth data to train models and the combinatorial complexity of introducing multiple alternative bases are hindering progress towards a goal of seamless basecalling from an extended alphabet of canonical and non-canonical bases. Downstream analyses, in particular, differential methylation, exploiting the phasing of base modifications, as well as visualisation, suffer from a dearth of tools.
The field of long-read transcriptomics is equally in its infancy. To date, the Iso-Seq pipeline has been used to build catalogues of transcripts in a range of species [128, 169, 170]. Nanopore reads-based transcriptomes are more recent [10, 171–173], and work is still needed to understand the characteristics of these data (e.g. coverage bias, sequence biases, reproducibility). Certain isoform assembly pipelines predict a large number of unannotated isoforms requiring validation and classification. Even accounting for artefacts and transcriptional noise, these early studies reveal an unexpectedly large diversity in isoforms. Benchmark data and studies will be required in addition to atlas-type sequencing efforts to generate high-quality transcript annotations that are more comprehensive than the current ones. Long reads theoretically confer huge advantages over short reads for transcript-level differential expression, however the low-level of replication and modest read counts obtained from long-read transcriptomic experiments are currently limiting. Until throughput increases and price decreases sufficiently, hybrid approaches that use long reads to define the isoforms expressed in the samples and short reads to get enough counts for well-powered differential expression may be successful; these do not yet exist.
Long-read sequencing technologies have already opened exciting avenues in genomics. Taking on the challenge of obtaining phased, accurate, and complete (including base modifications) genomes and transcriptomes that can be compared will require continued efforts in developing and benchmarking tools.
讨论
在写这篇文章的时候,大约花费1500美元,就可以获得30 g的≥99%精确的SMRT CCS(1个Sequel II 8M SMRT cell)或者50-150 g的噪音更大但可能更长的纳米孔读数(1个PromethION flow cell)。
虽然最初,长读测序可能对小(细菌)基因组的组装最有用,但最近的吞吐量和准确性的提高使更广泛的应用成为可能。
实际的生物聚合物,携带遗传信息现在可以测序全长或至少在数十到数百个碱基的片段,给我们一个更完整的基因组的照片(如telomere-to-telomere组件、结构变异、阶段性变化,表观遗传学,宏基因组)和转录组(例如同种型的多样性和数量、epitranscriptomics、聚腺苷酸化)。
这些进步的基础都是不断扩大的工具集合明确考虑长阅读的特点,特别是他们的出错率,有效、准确地执行任务,如预处理、纠错、对齐、组装、基础修改检测、量化、物种鉴定。
我们在long-read-tools.org数据库中整理了这些工具。
我们的人口调查揭示了长期阅读的分析工具的激增,这为基准测试的补充努力提供了一个令人信服的理由。
这个过程的关键是产生公开可用的基准数据集,其中地面真相是已知的,其特征是尽可能接近那些真实的生物数据集。
模拟、人工核酸合成等成绩单或vitro-methylated DNA,重新排序,验证努力将有助于建立一个地面实况的数组可以基准测试工具。
尽管技术、化学和数据格式的快速迭代,这些基准将鼓励最佳实践的出现。
复发性挑战读数据分析是可伸缩性。
例如,在基因组装配方面,Canu[69]可以为小的基因组生产出色的装配程序,但对于大的基因组则需要很长时间才能运行。
快速处理对于在应用程序中实现参数优化至关重要。
最近发布的wtdbg2[63]、TULIP[70]、Shasta[162]、Peregrine[163]、Flye[164]和Ra[165]汇编程序速度快了一个数级级,并迅速被采用。
类似地,对于长时间的映射读取,minimap2的速度,除了它的准确性之外,也为它的快速和广泛采用做出了贡献。
纳米极化[66]在装配校正和基修改检测中都很流行;
但是,它在大型数据集上运行速度很慢。
在f5c工具中重构call-methylation函数,极大地促进了对大基因组或数据集的处理[166]。
除了数据处理速度之外,可伸缩性还受到数据生成、存储和集成的影响。
纳米孔测序提供了最快的周转时间。
一旦DNA被提取,测序就在几分钟到几小时内进行,并且PromethION测序器提供可调节的高通量与单独寻址并行流细胞。
所有其他库准备过程都需要更多的劳力,测序可能需要等待池来完成一次运行,流单元需要连续运行而不是并行运行。
然而,原始纳米孔数据极其庞大(大约每基20字节),导致大型项目的大量IT成本。
SMRT电影不会保存以供以后重新设置碱基,每个碱基占用的序列和动力学信息更少,为3.5字节。
此外,融合了其他技术优势的混合方法,如光学测绘(Bionano, OpGen)和Hi-C,增加了基因组项目的成本和分析复杂性。
对于这些用户来说,手动数据集成是一个重要的瓶颈,但值得为此付出努力。
尽管SMRT和纳米孔测序平台的准确性都在提高,但纠错仍然是长时间读取分析管道中的重要一步。
发表的总成,省略小心纠错可能预测许多虚假的截短蛋白[167]。
混合错误校正利用了短读取的准确性,仍然优于长只读校正[60]。
现代的短读测序方案需要较小的输入量(有些甚至缩小到单个细胞),因此样本数量通常不会成为短读测序和长读测序结合的障碍。
通过改进非混合错误校正工具和/或长读测序精度,消除对短读的需求和更高的覆盖率,将降低基因组项目的成本、长度和复杂性。
长读测序所带来的表观遗传学/表转录组学方面的进展仍在发展中。
许多修饰,包括5mC,都不会对SMRT聚合酶的动力学产生足够的影响,从而在一个有用的灵敏度下被检测到(5mC需要250倍的覆盖)。
在这种情况下,软件的改进不太可能产生显著的收益,而改进化学排序可能是必需的[168]。
纳米孔测序似乎更适合广泛的基础上修改的检测(日期:5微克BrdU 6 ma),但缺乏地面实况数据训练模型和引入多个替代基地组合的复杂性阻碍进展的目标从一个扩展无缝basecalling字母的规范和非规范基地。
下游分析,特别是差异甲基化,开发阶段的碱基修饰,以及可视化,遭受工具的缺乏。
长期阅读的转录组学领域同样处于起步阶段。
迄今为止,isoo - seq管道已被用于构建一系列物种的转录本目录[128,169,170]。
基于纳米孔读取的转录组是最近才出现的[10,171 - 173],仍然需要工作来了解这些数据的特征(例如覆盖偏差、序列偏差、重现性)。
某些同种型装配管道预测大量未经亚型要求验证和分类。
甚至占文物和转录噪音,这些早期的研究揭示出意外大亚型的多样性。
除了阿特拉斯类型的测序工作之外,还需要基准数据和研究,以生成比现有的更全面的高质量转录文本注释。
从理论上讲,对于转录水平的差异表达来说,长读比短读具有巨大的优势,然而,从长读的转录组实验中获得的低水平的复制和适度的阅读计数目前受到限制。
在吞吐量增加和价格充分下降之前,使用长读来定义样品中表达的异构体,使用短读来获得足够的计数来实现高效的差异表达的混合方法可能是成功的;
这些还不存在。
长期以来,测序技术已经在基因组学领域开辟了令人兴奋的道路。
要想获得阶段性的、准确的、完整的(包括碱基修饰)基因组和转录组,从而能够进行比较,需要继续努力开发和基准测试工具。