从1977年Sanger发明“双脱氧链终止法”DNA测序技术起,基因组神秘的面纱一点一点的被揭露。
从小至几千碱基的噬菌体基因组到数百万碱基的细菌基因组,再到三十亿碱基的人类基因组,每一步都值得记录在人类探索自然、认识自身的篇章中。
而这些成果背后的重要一环——基因组组装,无疑是一个在研究中足够”美”的问题:既足够简明,可用短短的一段话来描述;又足够深刻,值得数十年的持续研究。
Part 1.白云生处

上面是计算机学家Staden关于序列拼接的描述,从中可以引申出我们如今经常使用的几个术语:reads/overlap/contig。
对于序列拼接的概念Staden进行了简明的定义:通过读取片段(reads)间的连接关系(overlap)构建出更长的连续性片段(contig)。
更进一步的组装研究中,序列拼接问题被转化为图论中的路径寻找问题:以点(node)代表测序序列,以边(edge)代表连接关系,以路径(path)代表的图上点的定向行走(walk)。
这里面,最有代表性的两种构图方式即string图和de Brujin图。下面的两张图,非常好的阐述了string图和de Brujin图在基因组组装中的应用原理。
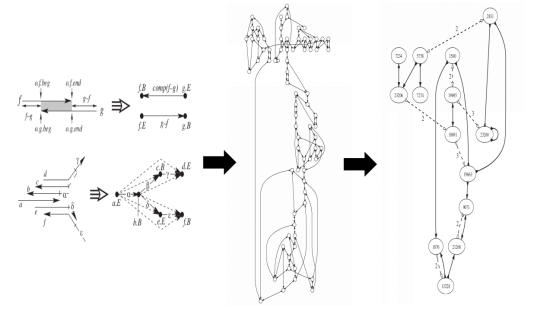
图1. Strings Graph in genome assembly[2]
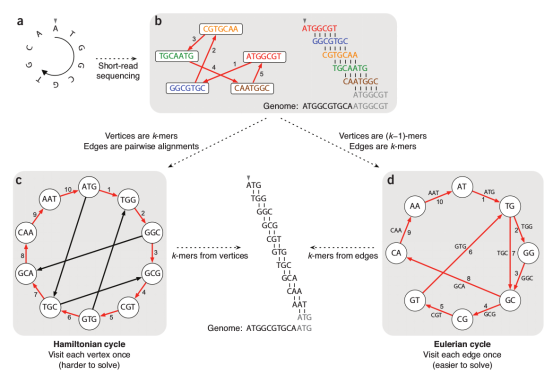
图2. De Brujin Graph in genome assembly[3]
Part 2.一往而深
提起基因组,最广为人知的应该是人类基因组计划了,2001年公布的人类基因组是这一计划的里程碑事件。
其中,大放光彩的Celera Assembler也成为基因组组装的”初代机“[4],whole genome shotgun的测序策略结合Overlap Layout Consensus的组装策略,攻克了基因组学研究上的第一座高峰。
但是一代测序由于高昂的测序成本以及较低的测序通量,限制了其在更多、更大规模的基因组学研究中的应用。
随着二代高通量测序的应运而生,全基因组测序才成为科研人员广泛使用的工具。以2005年出现的454测序仪和2008年出现的illumina测序仪为代表,短读长、高通量的测序数据成为主流。
而对于基因组组装而言,与之而来的却是短至几十碱基的测序片段带来的拼接困境。
为此,研究人员发明了不同的文库构建方法,以及改变了序列拼接的算法。高深度+多文库的双端测序策略结合de Brujin图的组装策略,成为新一代的组装标杆。
在这一风起云涌的时代,华大基因以其SOAPdenovo[5],以快打慢,打下了一片大大的江山(大雾)。
所谓一代版本一代神,虽然通过二代测序绘制了多物种的基因组草图,但整体的连续性和完整性上仍存在较大不足。
随着三代单分子测序技术的出现,又再次焕发了OLC组装策略的新春。
基于Celera Assembler,研究人员适应三代测序数据形成了Hierarchical Genome Assembly Process(HGAP)的先纠错再组装的策略[6]。
而二代测序并没有因此退出组装舞台,采用巧妙的文库构建方法如全基因组染色体构象捕获测序技术(Hi-C)、在DNA片段上加入高通量的barcode标签测序技术(10X)等,能够进一步对基因组进行完善升级,甚至使组装结果达到染色体水平。
说起来,最初接触组装时深入研究的就是Celera Assembler,当时还是三代测序出现之初的7.0版本,见证了诸多版本的更新,不得不说,开发人员确实是一往而深(大雾),当为吾辈楷模。
Part 3.沧海云帆
组装的终极目标是得到一个没有间隙(gap)的、单倍体精度的组装结果,但是目前为止,还没有一个高等动植物的基因组实现这样的目标。即使是研究最完善的人类基因组,目前仍存在800余个gaps。
但是这个目标的实现距离我们已经是触目可及了:测序技术不断发展,一代、二代、三代、光学等数据优势互补,使组装如虎添翼;建库方法不断改进,Hi-C、10X等方法画龙点睛,助组装锦上添花。
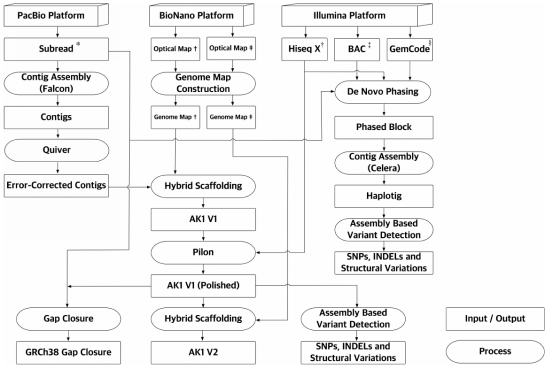
图3. 测序组装策略[7]
那么,基因组组装的未来是什么呢?
首先作为承载组学信息的基石,完整且准确的基因组是整合越来越多测序信息不可缺少的一环,这其中,组装必不可少.
其次,作为研究人员,最想看到的是没有组装!一条DNA从头测到尾,0 gap,不组装,测出即用。
有可能实现吗?让我们一起拭目以待吧。
参考文献
1. Adam M. Phillippy. New advances in sequence assembly.
2. Eugene W. Myers. The fragment assembly string graph.
3. Phillip Compeau. How to apply de Bruijn graphs to genome assembly.
4. Venter et al. The Sequence of the Human Genome.
5. Luo R, Liu B, Xie Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler.
6. Chin CS, Alexander DH, Marks P, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data.
7. Seo J-S, et al. De novo assembly and phasing of a Korean human genome.