http://note.youdao.com/share/?id=312fa04209cb87f7674de9a9544f329a&type=note#/
https://davetang.org/wiki/tiki-index.php?page=SAM
编辑距离Edit Distance:
从字符串a变到字符串b,所需要的最少的操作步骤(插入I,删除D,更改)为两个字符串之间的编辑距离。这也是sam文档中对NM这个tag的定义。编辑距离是对两个字符串相似度的度量(参见文章:Edit Distance http://www.cnblogs.com/lihaozy/archive/2012/12/31/2840152.html)
举个例子:两个字符串“eeba”和“abca”的编辑距离是多少?
根据定义,通过三个步骤:1.将e变为a 2.删除e 3.添加c,我们可以将“eeba”变为“abca”。所以,“eeba”和“abca”之间的编辑距离为3

1、如何通过bam文件统计比对的indel和mismatch信息
92S59M8I17M1D6M1D67M
(1)CIGAR 的格式:操作长度 + 相应的操作符。第6列。
常用的操作符有3个M for match or mismatch, I for insertion and D for deletion;此外还有一些扩展的操作符去描述 clipping, padding and splicing。(注:目前只 在blasr比对结果中见过=和X)
M(匹配比对,包含match和mismatch),=(纯match),X(纯mismatch),
I(插入到参考序列中),
D(从参考序列中删除),
N、(从参考序列中跳过)
S、Soft clip on the read (剪切掉的序列还在 in <seq>)
H、Hard clip on the read (剪切掉的序列不在 in <seq>)
P、silent deletion from the padded reference sequence
(2) cigar字段可以统计indel信息。
(3) cigar字段无法统计mismatch,这个时候就可以用到NM tag了,mismatch = NM – I - D = 25 – 8 – 1 – 1 = 15
2、Optional fields 的格式: <TAG>:<VTYPE>:<VALUE>。第12列
VTYPE:
| Type | Description |
| A | Printable character |
| i | Signed 32-bin interger |
| f | Single-precision float number |
| Z | Printable string |
| H | Hex string (high nybble first) |
TAG:
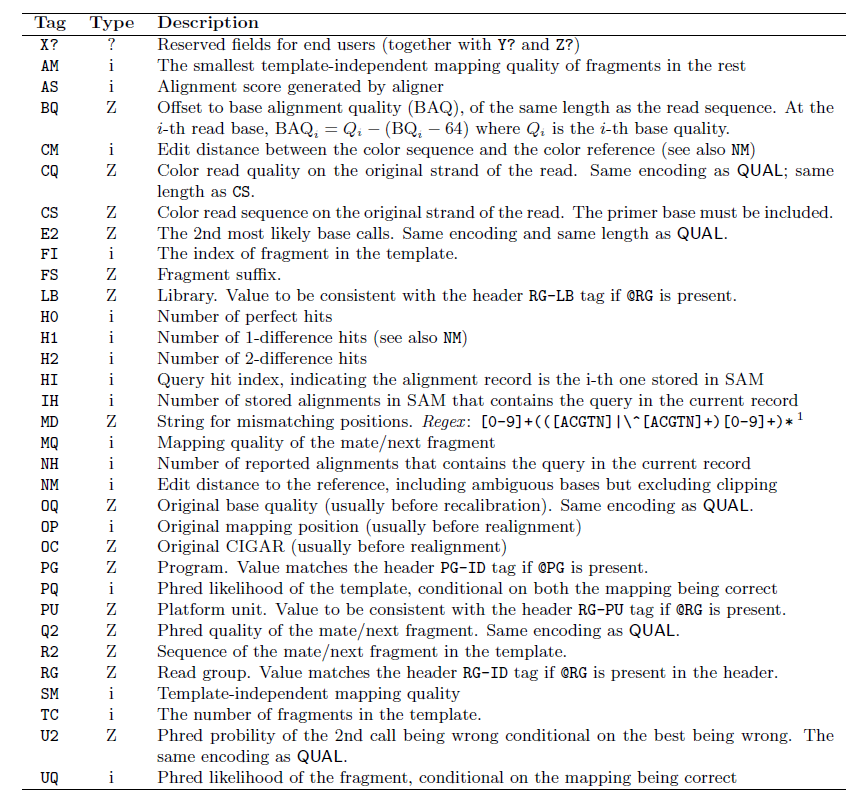
AS:i:<N> Alignment score.可以为负的,在local下可以为正的。 只有当Align≥1 time才出
XS:i:<N> Alignment score for second-best alignment. 当Align>1 time出现
YS:i:<N> Alignment score for opposite mate in the paired-end alignment. 当该read是双末端测序中的一条时出现
XN:i:<N> The number of ambiguous bases in the reference covering this alignment.(推测是指不知道错配发生在哪个位置,推测是针对于插入和缺失,待查证)
XM:i:<N> 错配碱基的数目
XO:i:<N> The number of gap opens(针对于比对中的插入和缺失)
XG:i:<N> The number of gap extensions(针对于比对中的插入和缺失)
NM:i:<N> The edit distance(read string转换成reference string需要的最少核苷酸的edits:插入/缺失/替换)
YF:Z:<S> 该reads被过滤掉的原因。可能为LN(错配数太多,待查证)、NS(read中包含N或者.)、SC(match bonus低于设定的阈值)、QC(failing quality control,待证)
YT:Z:<S> 值为UU表示不是pair中一部分(单末端?)、CP(是pair且可以完美匹配) DP(是pair但不能很好的匹配)、UP(是pair但是无法比对到参考序列上)
MD:Z:<S> 比对上的错配碱基的字符串表示
3、FLAG 第2列
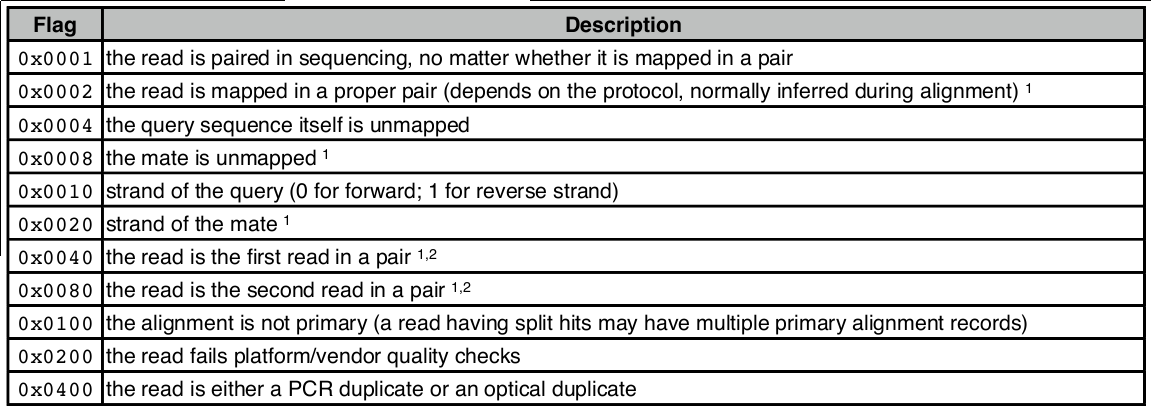
| 1 | the read is paired in sequencing, no matter whether it is mapped in a pair |
| 1 | the read is mapped in a proper pair |
| 0 | not unmapped |
| 0 | mate is not unmapped |
| 0 | forward strand |
| 1 | mate strand is negative |
| 0 | the read is not the first read in a pair |
| 1 | the read is the second read in a pair |
4、 alignments的类型
SAM可以存储 clipped alignments, spliced alignments, multi-part alignments, padded alignments and alignments in colour space
11个必须的字段(mandatory fields)和一个可选的字段,字段之间用tag分割
<QNAME> <FLAG> <RNAME> <POS> <MAPQ> <CIGAR> <MRNM> <MPOS> <ISIZE> <SEQ> <QUAL> [<TAG>:<VTYPE>:<VALUE> [...]]
1. QNAME,比对片段的(template)的编号;
2. FLAG,位标识,template mapping情况的数字表示,每一个数字代表一种比对情况,这里的值是符合情况的数字相加总和;
3. RNAME,参考序列的编号,如果注释中对SQ-SN进行了定义,这里必须和其保持一致,另外对于没有mapping上的序列,这里是’*‘;
4. POS,比对上的位置,注意是从1开始计数,没有比对上,此处为0;
5. MAPQ,mappint的质量;
6. CIGAR,简要比对信息表达式(Compact Idiosyncratic Gapped Alignment Report),其以参考序列为基础,使用数字加字母表示比对结果,比如3S6M1P1I4M,前三个碱 基被剪切去除了,然后6个比对上了,然后打开了一个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的;
7. RNEXT,下一个片段比对上的参考序列的编号,没有另外的片段,这里是’*‘,同一个片段,用’=‘;
8. PNEXT,下一个片段比对上的位置,如果不可用,此处为0;
9. TLEN,Template的长度,最左边得为正,最右边的为负,中间的不用定义正负,不分区段(single-segment)的比对上,或者不可用时,此处为0;
10、SEQ,序列片段的序列信息,如果不存储此类信息,此处为’*‘,注意CIGAR中M/I/S/=/X对应数字的和要等于序列长度;
11.QUAL,序列的质量信息,格式同FASTQ一样。
12、可选字段(optional fields),格式如:TAG:TYPE:VALUE,其中TAG有两个大写字母组成,每个TAG代表一类信息,每一行一个TAG只能出现一次,TYPE表示TAG对应值 的类型,可以是字符串、整数、字节、数组等。
(1)Clipped alignment
REF: AGCTAGCATCGTGTCGCCCGTCTAGCATACGCATGATCGACTGTCAGCTAGTCAGACTAGTCGATCGATGTG
READ: gggGTGTAACC-GACTAGgggg
read中大写字母表示与参考基因匹配,小写字母表示read的剪掉部分。- 表示与参考基因组相比read缺失的碱基中 。例子是 3S8M1D6M4S ( 3 soft, 8 match, 1 deletion, 6 match and 4 soft).
(2)Spliced alignment
在 cDNA-to-genome的比对中, 为了区分内含子(...),外显子的缺失(---)。通过引入操作符“”N“来代表在参考序列上长的skip,(we may want to distinguish introns from deletions in exons.)
REF: AGCTAGCATCGTGTCGCCCGTCTAGCATACGCATGATCGACTGTCAGCTAGTCAGACTAGTCGATCGATGTG READ: GTGTAACCC................................TCAGAATA
'...' 表示 intron. 这个比对的CIGAR是 : 9M32N8M.
(3)Multi-part alignment
One query sequence may be aligned to multiple places on the reference genome, either with or without overlaps. In SAM, we keep multiple hits as multiple alignment records. To avoid presenting the full query sequence multiple times for non-overlapping hits, we introduce operation 'H' to describe hard clipped alignment. Hard clipping (H) is similar to soft clipping (S). They are different in that hard clipped subsequence is not present in the alignment record. The example alignment in "clipped alignment" can also be represented with CIGAR: 3H8M1D6M4H, but in this case, the sequence stored in SAM is "GTGTAACCGACTAG", instead of "GGGGTGTAACCGACTAGGGGG" if soft clipping is in use.
(4)Padded alignment
Most sequence aligners only give the sequences inserted to the reference genome, but do not present how these inserted sequences are aligned against
each other. Alignment with inserted sequences fully aligned is called padded alignment. Padded alignment is always produced by de novo assemblers and
is important for an alignment viewer to display the alignment properly. To store padded alignment, we introduce operation 'P' which can be considered
as a silent deletion from padded reference sequence. In the following example, GA on READ1 and A on READ2 are inserted to the reference. With unpadded
CIGAR, we would not be able to distinguish the following padded multi-alignments:
REF: CACGATCA**GACCGATACGTCCGA READ1: CGATCAGAGACCGATA READ2: ATCA*AGACCGATAC READ3: GATCA**GACCG The padded CIGAR are different: READ1: 6M2I8M READ2: 4M1P1I9M READ3: 5M2P5M
(5)Alignments in colour space
Colour alignments are stored as normal nucleotide alignments with additional tags describing the raw colour sequences, qualities and colour-specific
properties.