参考博客:https://blog.csdn.net/weixin_43569478/article/details/83745105
需要创建一个cls文件
cls文件格式
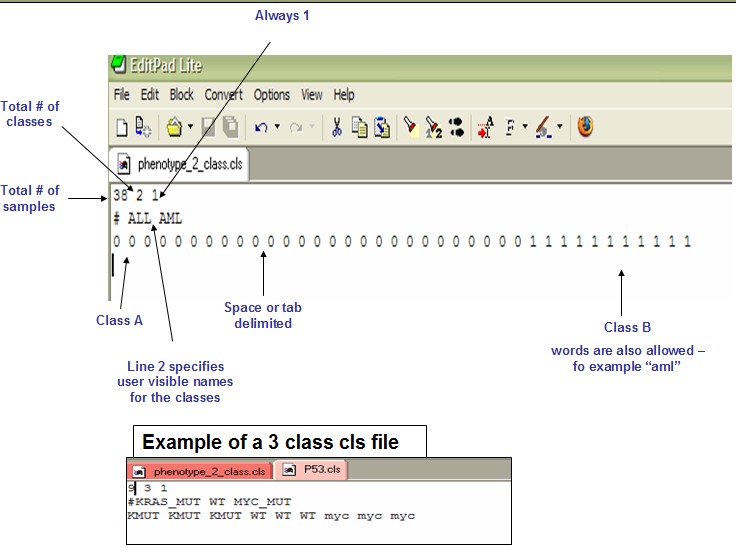
10 2 1
#AdA3_0h AdA3_16h
AdA3_0h AdA3_0h AdA3_0h AdA3_0h AdA3_0h AdA3_0h AdA3_16h AdA3_16h AdA3_16h AdA3_16h
-
第一行的三个数字分别表示10个样本,2个分组,总是设置为1
-
第二行为组的名称
-
第三行为组的重复个数,也就是每个组有几个重复就写几次
另外需要一个mmu.kegg_optimal.symbol.gmt
这个文件可以在GSEA官网下载也可以自己制作一个相应的格式,如果自己制作需要有kegg注释的结果
-
对差异表达的结果进行gsea分析
java -Xmx2048m -cp /biosoftware/gsea-3.0.jar xtools.gsea.Gsea -res Flox.HFHC.16W_vs_CKO.HFHC.16W.fpkm.txt -cls Flox.HFHC.16W_vs_CKO.HFHC.16W.class.cls -gmx mmu.kegg_optimal.symbol.gmt -out Flox.HFHC.16W_vs_CKO.HFHC.16W -rpt_label Flox.HFHC.16W_vs_CKO.HFHC.16W -collapse false -mode Max_probe -norm meandiv -nperm 1000 -permute gene_set-rnd_type no_balance -scoring_scheme weighted -rpt_label my_analysis -metric Signal2Noise -sort real-order descending -include_only_symbols true -make_sets true -median false -num 100 -plot_top_x 500-rnd_seed timestamp -save_rnd_lists false -set_max 500 -set_min 15 -zip_report false -gui false
- GSEA的结果文件的理解
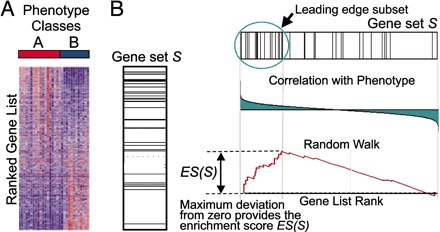
这是一张原理图,GSEA的输入是一个基因表达量矩阵,其中的样本分成了A和B两组,首先对所有基因进行排序,在之前的文章中也有提到排序的标准,这里简单理解就是foldchange, 用来表示基因在两组间表达量的变化趋势。排序之后的基因列表其顶部可以看做是上调的差异基因,其底部是下调的差异基因
GSEA分析的是一个基因集下的所有基因是否在这个排序列表的顶部或者底部富集,如果在顶部富集,我们可以说,从总体上看,该基因集是上调趋势,反之,如果在底部富集,则是下调趋势。
GASE给出了一个汇总的html页面。对于富集结果,根据上调还是下调分成了两个部分,对应两个分组,示例如下:

在每个组别下富集到的基因集,从总体上看,其表达量在该组中高表达。点击enrichment results in html,可以在网页查看富集的结果,示例如下
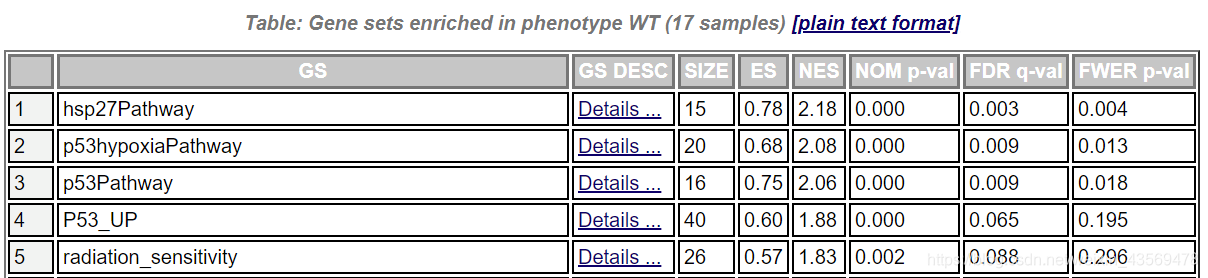
GS为基因集的名字,SIZE代表该基因集下的基因总数,ES代表Enrichment score, NES代表归一化后的Enrichment score, NOM p-val代表pvalue,表征富集结果的可信度,FDR q-val代表qvalue, 是多重假设检验矫正后的p值,注意GSEA采用pvalue < 5%, qvalue < 25% 对结果进行过滤。
点击GS DESC可以跳转到每个基因集详细结果页面,示例如下
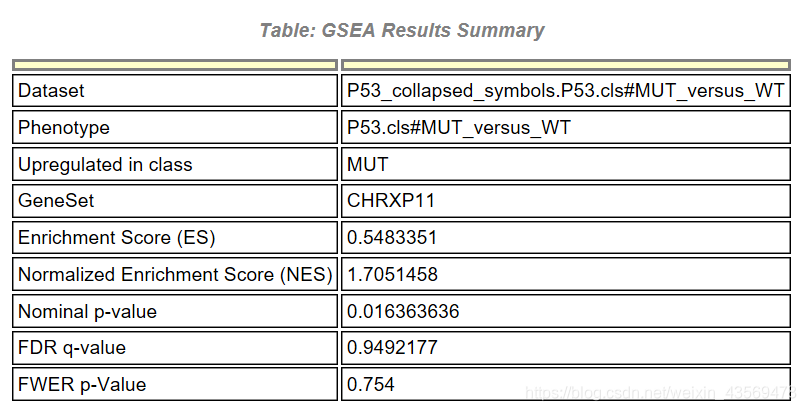
首先是一个汇总的结果,Upregulated in class说明该基因集在MUT这组中高表达,其他信息和之前介绍的一样,除此之外,还有一个详细的表格,示例如下
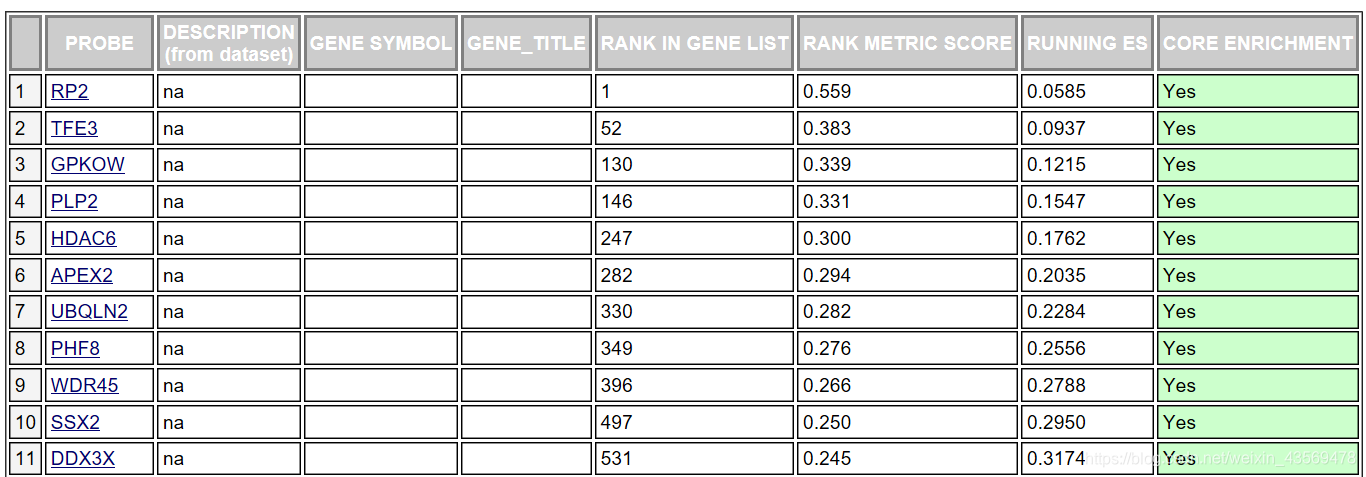
对于该基因集下的每个基因给出了详细的统计信息,RANK IN GENE LIST代表该基因在排序号的列表中的位置, RANK METRIC SCORE代表该基因排序量的值,比如foldchange值,RUNNIG ES代表累计的Enrichment score, CORE ENRICHMENT代表是否属于核心基因,即对该基因集的Enerchment score做出了主要贡献的基因。
这个表格中的数据对应下面这张图
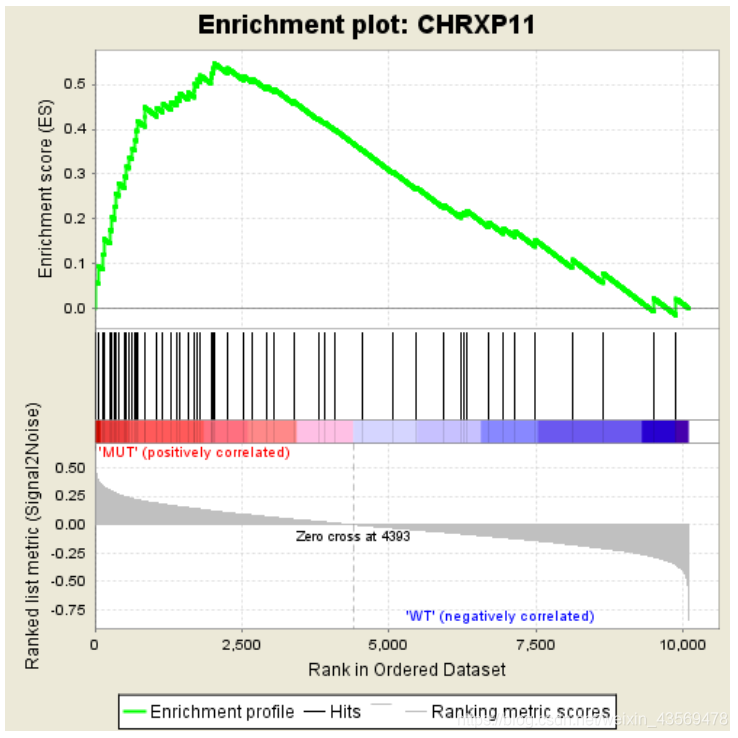
分成3个部分,第一部分为基因Enrichment Score的折线图,横轴为该基因下的每个基因,纵轴为对应的Running ES, 在折线图中有个峰值,该峰值就是这个基因集的Enrichemnt score,峰值之前的基因就是该基因集下的核心基因。
第二部分为hit,用线条标记位于该基因集下的基因,第三部分为所有基因的rank值分布图, 默认采用Signal2Noise算法,对应了纵轴的标题。
从该图中可以看出,这个基因集是在MUT这一组高表达的,下面是一个在另一组组中高表达的示例
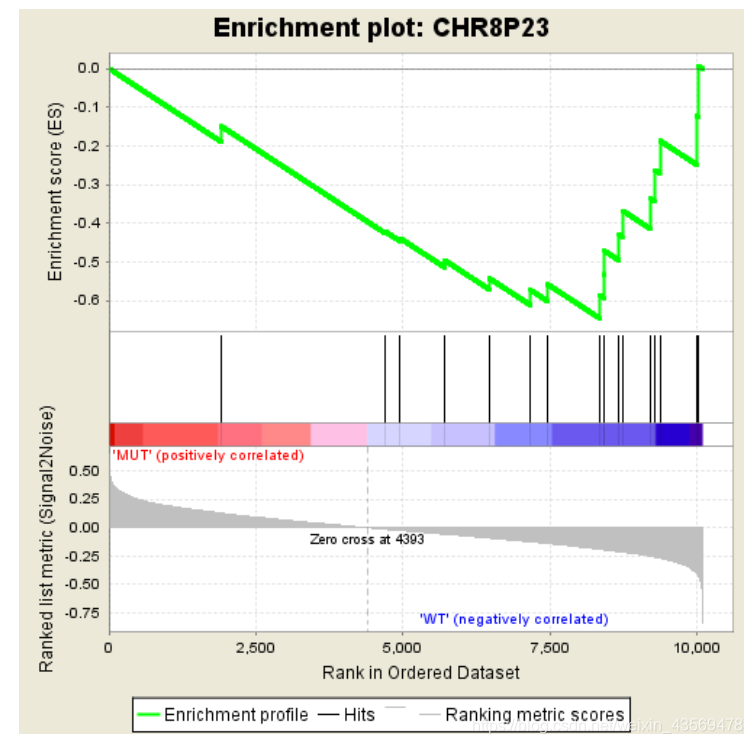
可以看到,其Enrichment score值全部为负数,对应的在其峰值右侧的基因为该基因集下的核心基因。
除此之外,还有一种热图,示例如下
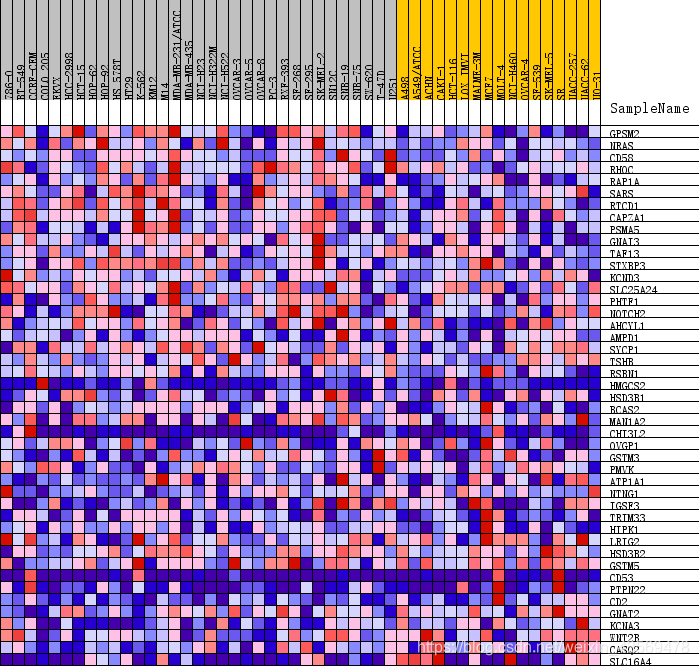
这张热图展示的是位于该基因集下的基因在所有样本中表达量的分布,其中每一列代表一个样本。每一行代表一个基因,基因表达量从低到高,颜色从蓝色过渡到红色。
在总的html页面中,还给出了如下信息
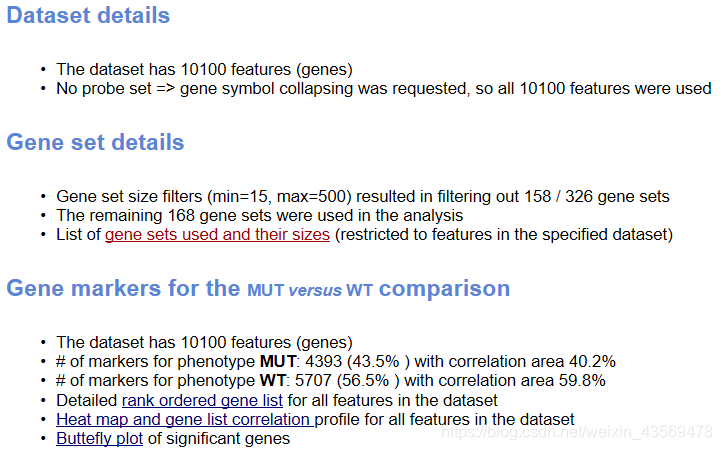
Dataset details给出了基因总数,Gene Set details给出了基因集的信息,注意软件默认根据基因集包含的基因个数是先对基因集进行过滤,最小15个,最大500个基因,过滤掉了158个基因集,剩余的168个基因集用于分析。
Gene markers给出了排序之后的基因列表和对应的统计量rank ordered gene list,根据排序的统计量,将基因分成了两部分,对应在每一组中高表达。heatmap and gene list包含了所有基因表达量的热图和排序值的分布图,示意如下
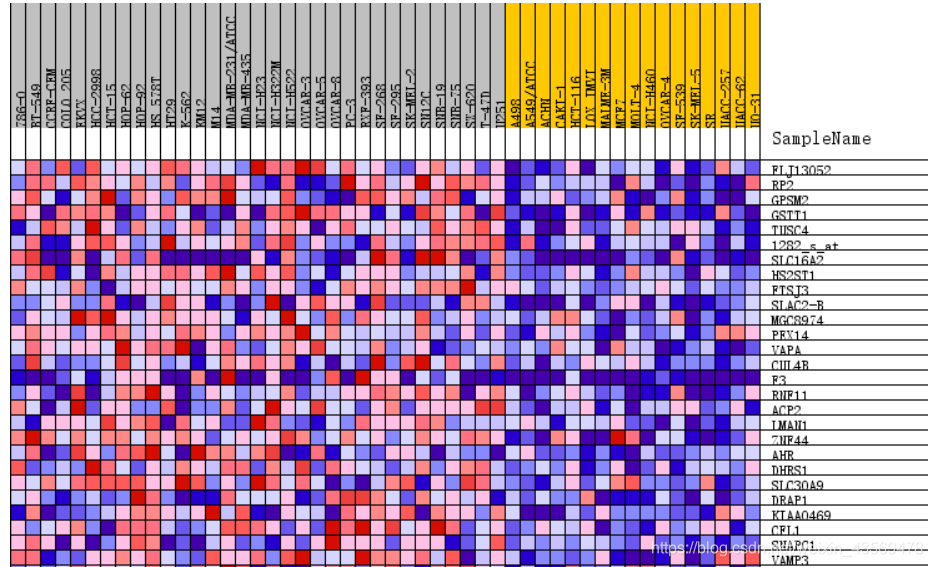
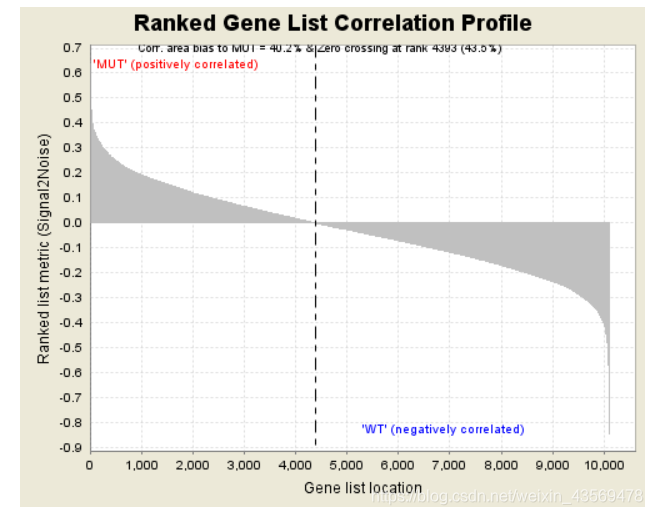
热图由于基因太多,截取了部分,排序值的分布图其实就是每个基因集的Enrichment plot中的第三部分