对分子进行聚类分析,首先必须要考虑的是其描述符的问题,分子描述符通常是非常高维的,必须对其进行降维才好继续后面的分析,特别分子量特别大的时候。常用的降维手段有PCA,TSNE和UMAP.一说,TSNE用于可视化.
聚类的方法有许多,比如k-means,层次聚类. 但是这两个一个需要定义k,一个需要定义阈值,这样需要试错法合理进行着两个量的设置,不是很方便. 因而,我选择使用HDBSCAN,一个基于数据密度的聚类算法,参考文献如下:
https://link.springer.com/chapter/10.1007%2F978-3-642-37456-2_14
我选择使用 HDBSCAN.
先导入所需要的各种库. 后面还会使用到 mlinsght 库.
%matplotlib inline
import os
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
from rdkit import Chem, DataStructs
from rdkit.Chem import AllChem, Draw
from rdkit import RDLogger
from sklearn.model_selection import train_test_split
from sklearn.gaussian_process import GaussianProcessRegressor
from sklearn.ensemble import RandomForestRegressor
from sklearn.manifold import TSNE
from mlinsights.mlmodel import PredictableTSNE
from hdbscan import HDBSCAN
sns.set_context('poster')
sns.set_style('white')
sns.set_color_codes()
plot_kwds = {'alpha' : 0.5, 's' : 80, 'linewidths':0}
RDLogger.DisableLog('rdApp.*')
seed = 794
HDBSCAN需要接受一个衡量分子间距离或者相似度的参数,可以自定义,这里我定义了tanimoto-dist函数,使用方法如下面的代码所示:
使用的时候发现自定义的tanimoto速度太慢了。。。。
oxrs = [("CHEMBL3098111", "Merck" ), ("CHEMBL3867477", "Merck" ), ("CHEMBL2380240", "Rottapharm" ),
("CHEMBL3352684", "Merck" ), ("CHEMBL3769367", "Merck" ), ("CHEMBL3526050", "Actelion" ),
("CHEMBL3112474", "Actelion" ), ("CHEMBL3739366", "Heptares" ), ("CHEMBL3739395", "Actelion" ),
("CHEMBL3351489", "Eisai" )]
fps = []
docs = []
companies = []
mol_list = []
for cid, company in oxrs:
sdf_file = os.path.join("data", cid + ".sdf")
mols = Chem.SDMolSupplier(sdf_file)
for mol in mols:
if mol is not None:
mol_list.append(mol)
fp = AllChem.GetMorganFingerprintAsBitVect(mol, 2)
arr = np.zeros((1,))
DataStructs.ConvertToNumpyArray(fp, arr)
docs.append(cid)
companies.append(company)
fps.append(arr)
fps = np.array(fps)
companies = np.array(companies)
def tanimoto_dist(ar1, ar2): # 接收两个numpy 数组
a = np.dot(ar1, ar2)
b = ar1 + ar2 - ar1*ar2
return 1 - a/np.sum(b)
# 实例化HDBSCAN类
clusterer = HDBSCAN(algorithm='best',
min_samples=5,
metric='pyfunc',
func=tanimoto_dist)
clusterer.fit(fps)
docs = np.array(docs)
trainIDX, testIDX = train_test_split(range(len(fps)), random_state=seed)
接下来我们来对数据进行可视化,使用到了tsne技术,下面两图的分子分别以company作为颜色标签和HDBSACA聚类结果作为颜色标签.
tsne = TSNE(random_state=seed)
res = tsne.fit_transform(fps)
plt.clf()
plt.figure(figsize=(12, 6))
sns.scatterplot(res[:,0], res[:,1], hue=companies, **plot_kwds)
plt.clf()
plt.figure(figsize=(12, 6))
palette = sns.color_palette()
cluster_colors = [sns.desaturate(palette[col], sat)
if col >= 0 else (0.5, 0.5, 0.5) for col, sat in zip(clusterer.labels_, clusterer.probabilities_)]
plt.scatter(res[:,0], res[:,1], c=cluster_colors, **plot_kwds)
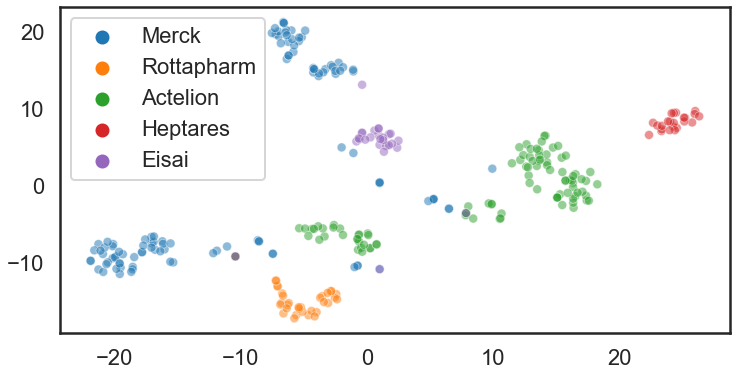
coloured with company name
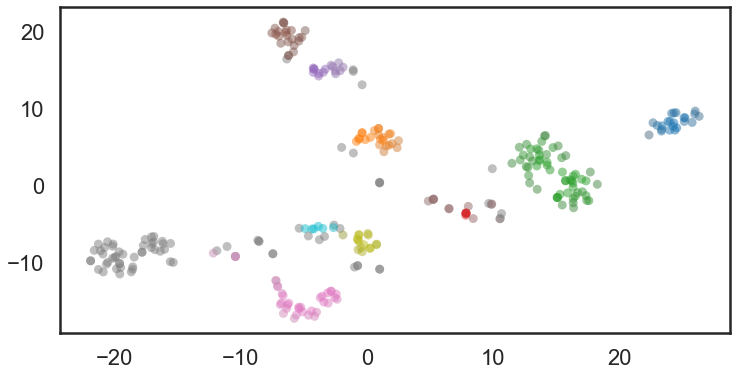
coloured with hdbscan label.
图中灰色的是没有标签的数据。结果表明HDBSCAN还是聚的不错的。
映射新化合物到已定义的化学空间
下一个主题是如何在预定的化学空间中绘制新化合物。 从理论上讲,PCA和tSNE不能将新数据添加到预定义的潜在分布空间。 如果用户想添加新数据,则需要重新计算。 但是我们想将新设计的化合物映射到当前的化学空间。 嗯... 怎么做?
答案是: mlinghts. 这个库包含 PredictableTSNE 类.从名字上就不难看出它是干啥的了。 这个类接收一个TSNE实例和一个用于预测的估计器.
在下面的例子中,我们使用 random forest 和 GP回归器.
trainFP = [fps[i] for i in trainIDX]
train_mol = [mol_list[i] for i in trainIDX]
testFP = [fps[i] for i in testIDX]
test_mol = [mol_list[i] for i in testIDX]
allFP = trainFP + testFP
tsne_ref = TSNE(random_state=seed)
res = tsne_ref.fit_transform(allFP)
plt.clf()
plt.figure(figsize=(12, 6))
sns.scatterplot(res[:,0],
res[:,1],
hue=['train' for i in range(len(trainFP))] + ['test' for i in range(len(testFP))])
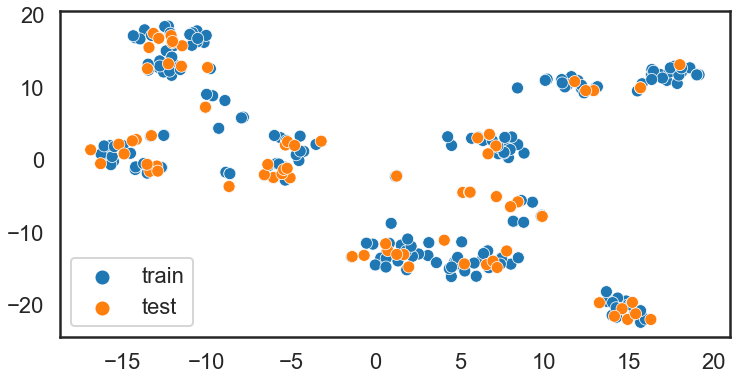
蓝色的是训练数据,橙色是测试数据.
使用predictiveTSNE 可以预测新化合物在化学空间的所在.下面的例子分别为RF和GP.
rfr = RandomForestRegressor(random_state=seed)
tsne1 = TSNE(random_state=seed)
pred_tsne_rfr = PredictableTSNE(transformer=tsne1,
estimator=rfr,
keep_tsne_outputs=True)
pred_tsne_rfr.fit(trainFP, list(range(len(trainFP))))
pred1 = pred_tsne_rfr.transform(testFP)
plt.clf()
plt.figure(figsize=(12, 6))
plt.scatter(pred_tsne_rfr.tsne_outputs_[:,0],
pred_tsne_rfr.tsne_outputs_[:,1],
c='blue',
alpha=0.5)
plt.scatter(pred1[:,0],
pred1[:,1],
c='red',
alpha=0.5)
gbr = GaussianProcessRegressor(random_state=seed)
tsne2 = TSNE(random_state=seed)
pred_tsne_gbr = PredictableTSNE(transformer=tsne2, estimator=gbr, keep_tsne_outputs=True)
pred_tsne_gbr.fit(trainFP, list(range(len(trainFP)))pred2 = pred_tsne_gbr.transform(testFP)
plt.clf()
plt.figure(figsize=(12, 6))
plt.scatter(pred_tsne_gbr.tsne_outputs_[:,0], pred_tsne_gbr.tsne_outputs_[:,1], c='blue', alpha=0.5)
plt.scatter(pred2[:,0], pred2[:,1], c='red', alpha=0.5))
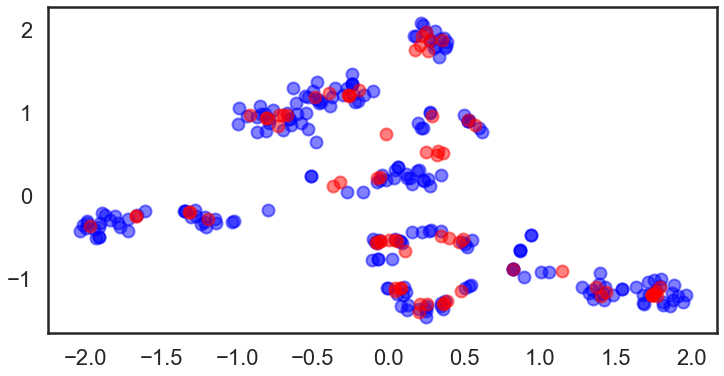
RF
GP
看起来RF的效果要好些.
预测算法对TSNE的性能有很大的影响。 这对于绘制新数据很有用,但存在设计误导的风险。
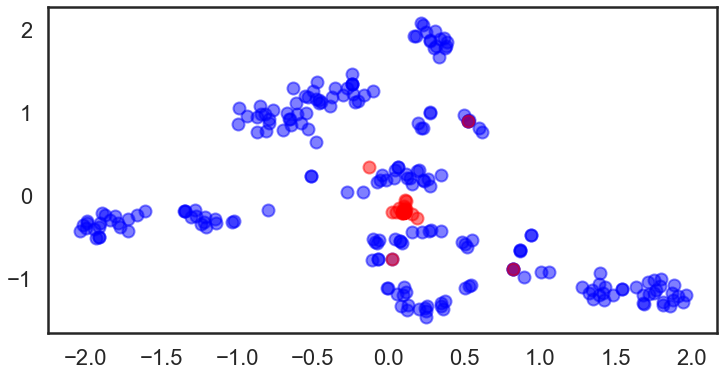
最后,检查一下聚类标签和preditiveTSNE 预测数据.
allmol = train_mol + test_mol
fps2 = []
clusterer2 = HDBSCAN(algorithm='best',
min_samples=5,
metric='pyfunc',
func=tanimoto_dist)
# calc fp
for mol in allmol:
fp = AllChem.GetMorganFingerprintAsBitVect(mol, 2)
arr = np.zeros((1,))
DataStructs.ConvertToNumpyArray(fp, arr)
fps2.append(arr)
fps2 = np.array(fps2)
# cluster
clusterer2.fit(fps2)
# plot
plt.clf()
plt.figure(figsize=(12, 6))
plt.scatter(pred_tsne_rfr.tsne_outputs_[:,0],
pred_tsne_rfr.tsne_outputs_[:,1],
c=clusterer2.labels_[:len(trainFP)],
alpha=0.5,
marker='x')
plt.scatter(pred1[:,0],
pred1[:,1],
c=clusterer2.labels_[len(trainFP):],
alpha=0.5, marker='o')
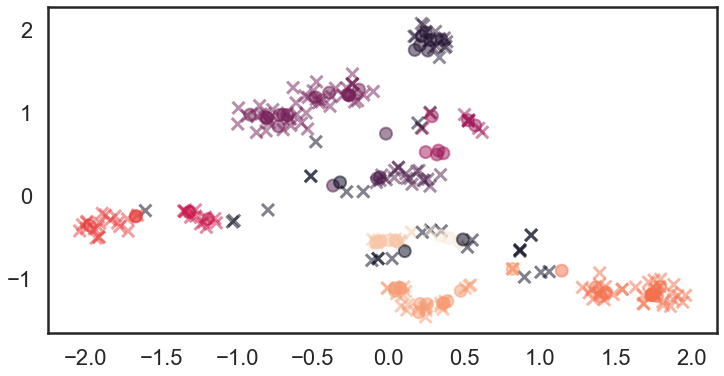
x是训练数据, o为测试数据, 颜色 :类别标签.
不仅训练数据而且具有相同颜色的测试数据也出现在几乎相同的区域中。 这表明该方法在这种情况下效果很好。