# Anaconda
一、modeller下载与安装
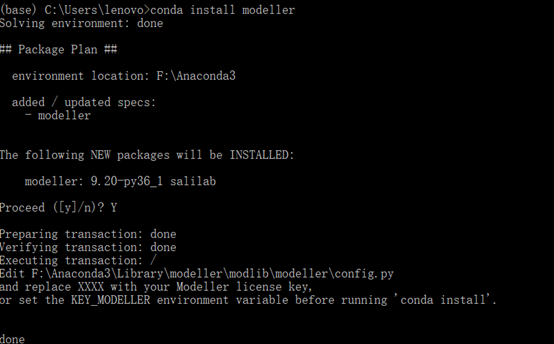
输入key码:MODELIRANJE

做下Basic Modeling 流程吧:

在python环境下运行下例子吧:(build_profile.py、compare.py、align2d.py、model-single.py、evaluate_model.py)

(这里还需要一个evaluate_template文件)

二、使用modeller对目的蛋白序列同源建模
>p1:dfra12
MNSESVRIYLVAAMGANRVIGNGPNIPWKIPGEQKIFRRLTEGKVVVMGRKTFESIGKPLPNRHTLVISRQANYRATGCVVVSTLSHAIALASELGNELYVAGGAEIYTLALPHAHGVFLSEVHQTFEGDAFFPMLNETEFELVSTETIQAVIPYTHSVYARRNG
1、添加额外的库:

Blast与PDB搜索,额外从公共数据库选两个相似蛋白来作为模版:2ZZA A 3JVX A(发现挑重复了于是我换了一个)

>P1;2ZZAA
structure:2ZZA: 1:A:
VIVSMIAALANNRVIGLDNKMPWHLPAELQLFKRATLGKPIVMGRNTFESIGRPLPGRLNIVLSRQTDYQPEGVTVVATLEDAVVAAGDVEELMIIGGATIYNQCLAAADRLYLTHIELTTEGDTWFPDYEQYNWQEIEHESYAADDKNPHNYRFSLLERVK
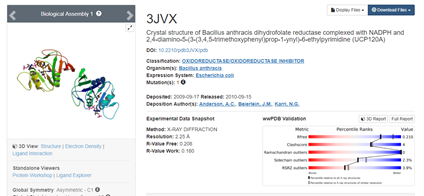
>3JVX:A|PDBID|CHAIN|SEQUENCE
HHHHHHMRVSFMVAMDENRVIGKDNNLPWRLPSELQYVKKTTMGHPLIMGRKNYEAIGRPLPGRRNIIVTRNEGYHVEGCEVAHSVEEVFELCKNEEEIFIFGGAQIYDLFLPYVDKLYITKIHHAFEGDTFFPEMDMTNWKEVFVEKGLTDEKNPYTYYYHVYEKQQ
将数据库中的模版序列按下面格式添加入:pdb_95.pir中:
2、运行build_profile.py:修改代码
aln.append(file='dfra12.ali',alignment_format='PIR',align_codes='ALL')行。

Selecting a template(模版)
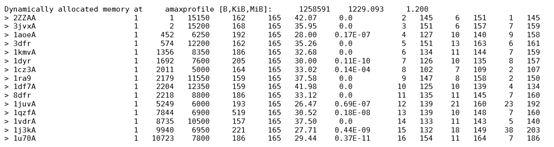
挑选E值为0,相似度大于35的序列:3dfr、1ra9、1df7A、1vdrA以及添加的2ZZAA、3jvxA,共五个模版,同时在PDB中下载对应的模版文件。
3、运行compare.py:
使用compare.py模版之间的结构和序列相似性。(修改对应的PDB)
输出结果如下:
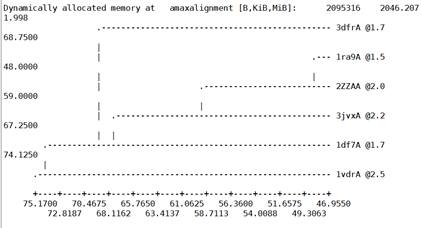
分析下:1ra9A与2ZZAA具有几乎相同的结构和序列,1ra9A有更好的晶体结构,选1ra9A。3dfrA与1df7A也可以选,它们也有相似的结构域,分辨率也不错。
4、运行align2d.py:
dfra12分别与模版比对(1ra9A、3dfrA、1df7A)
修改并运行程序:
得到两个文件(dfra12-1ra9A ali与pap格式文件),打开pap格式文件:

同理再运行两次:
3dfrA:

1df7A:

5、运行model-single.py对dfra12三维建模:
获得了dfra12-1ra9A类似的文件就可以进行三维建模了。(1ra9A、3dfrA、1df7A)——这里还可以再跑两轮,我就没跑了,重复的过程,所以对选择会有点偏差,但是足够建模。(最后我还是跑了下)
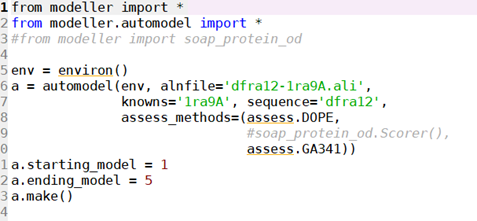
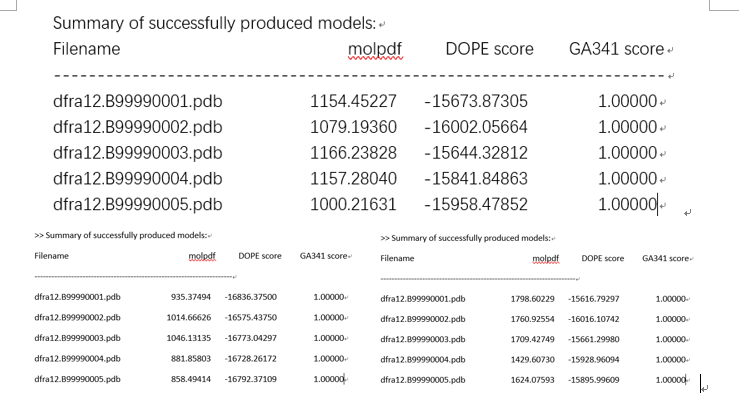
由此可以得出不同的pdb文件:选molpdf和DOPE scroe高的模型,这里选第三个dfra12.B99990001。(1df7A)
用Chimera打开模型:

6、模型评估:(evaluate_model、plot_profiles)选dB1评估
运行evaluate_model:

得到:dfra12.profile文件;score : -15616.977539
运行evaluate_template.py:
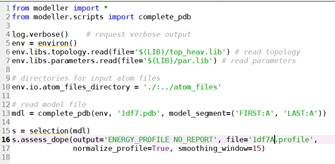
得到:1df7A.profile文件;score: -17980.896484
运行plot_profiles:函数块之后的代码截图如下,
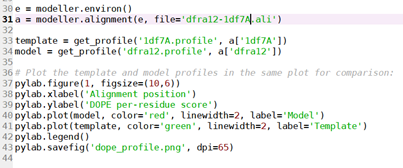
 (residue-alignment)
(residue-alignment)
相关文件:
链接:https://pan.baidu.com/s/1zKisFWJjtO2bdnxdO9Oncw
提取码:bfk3