suppa2继续测试,这个软件真的非常好用,速度很快、文档清晰、使用方便。
分析步骤小结:
- 用salmon快速比对,reference是转录本fasta文件,得到的是每个转录本的TPM;
- 根据gtf文件构建AS events,分类清晰,主流标准【science paper为证】;
- 根据suppa自己的算法,由TPM矩阵和AS events得到每个AS event的PSI矩阵;
算法清晰,就是TPM的比值,PSI也是比值
AS event阐述清晰,格式明确

举例:
# 一个AF event # ENSG00000011304;AF:chr19:797452:797505-799413:798428:798545-799413:+ # PSI的计算方法 # ENST00000394601 ENST00000394601,ENST00000587094
可视化:toolTesting/AS_and_isoforms_checking.ipynb
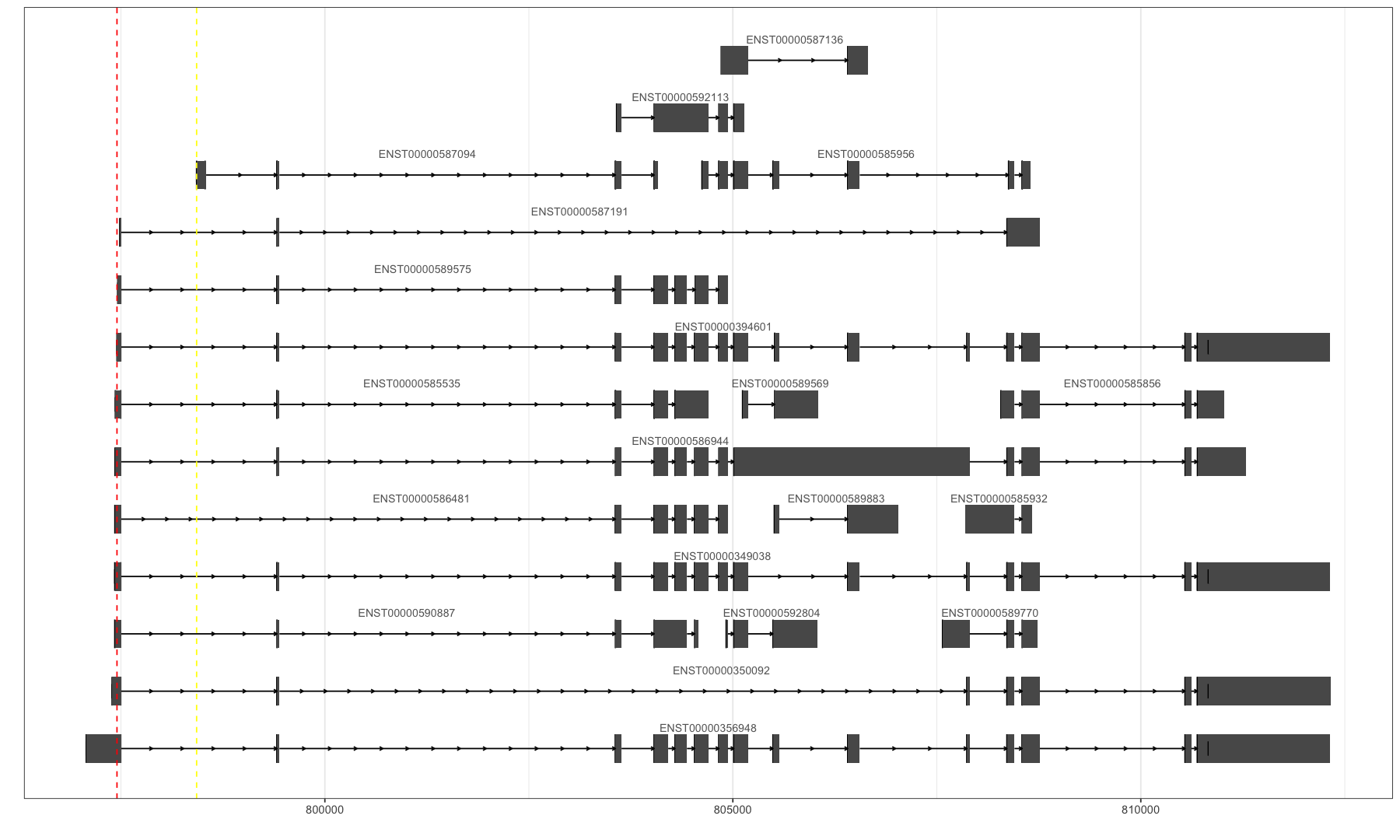
关于方向:
目前AS event里的skipping和exon都搞明白,没有歧义了,那方向呢?怎么确定哪个isoform是我计算的那个PSI的分子?【非常重要】
看AS event的定义,第一个就是分子:ENST00000394601 ENST00000394601,ENST00000587094
ENST00000394601特有的那个exon就是分子,在画kartoon图的时候应该放在下面。
ggplot画gene structure和alternative splicing | ggbio | GenomicFeatures
至此,AS分析的所有细节已经全部明确,感受一下数据之美!
在测试的一个工具:SUPPA2
https://github.com/comprna/SUPPA
测试代码:
salmon快速比对,得到exon TPM
source activate splicing salmon index -t gencode.v37.transcripts.fa -i gencode.v37.transcripts.salmon.index salmon index -t hg19_EnsenmblGenes_sequence_ensenmbl.fasta -i Ensembl_hg19_salmon_index
# source activate splicing
# hg38
index=~/references/SUPPA2/ref/GRCh38/gencode.v37.transcripts.salmon.index/
# hg19
# index=~/references/SUPPA2/ref/hg19/Ensembl_hg19_salmon_index/
for i in `ls merged.fastq/*.list0*`
#for i in `ls merged.fastq/{IMR90.list*,17C8.list*}`
do
echo $i &&
cut -d, -f2 $i | xargs zcat > ${i}_1.fastq &&
cut -d, -f3 $i | xargs zcat > ${i}_2.fastq &&
# gzip fastq/${i}_1.fastq fastq/${i}_2.fastq &&
~/softwares/anaconda3/envs/splicing/bin/salmon quant -i $index -l ISF --gcBias -1 ${i}_1.fastq -2 ${i}_2.fastq -p 10 -o ${i}_output &&
rm ${i}_1.fastq ${i}_2.fastq &&
echo "done"
done
suppa后续分析
#####################################################
# suppa analysis
~/project/scRNA-seq/rawData/smart-seq/analysis/suppa/
# index folder
~/references/SUPPA2/ref/hg19/Ensembl_hg19_salmon_index/
# prepare annotation of AS events
python ~/softwares/SUPPA/suppa.py generateEvents -i Homo_sapiens.GRCh37.75.formatted.gtf -o ensembl_hg19.events -e SE SS MX RI FL -f ioe
awk '
FNR==1 && NR!=1 { while (/^<header>/) getline; }
1 {print}
' *.ioe > ensembl_hg19.events.ioe
# merge TPM
python ~/softwares/SUPPA/multipleFieldSelection.py -i merged.fastq/*_output/quant.sf -k 1 -f 4 -o iso_tpm.txt
# format the rowname
# error: non-unique values when setting 'row.names': ‘ENSG00000000003.15’, ‘ENSG00000000005.6’
# modify ~/softwares/SUPPA/scripts/format_Ensembl_ids.R
# ids <- unlist(lapply(rownames(file),function(x)strsplit(x,"\|")[[1]][1]))
Rscript ~/softwares/SUPPA/scripts/format_Ensembl_ids.R iso_tpm.txt
# get PSI values
# hg19
python ~/softwares/SUPPA/suppa.py psiPerEvent -i ~/references/SUPPA2/ref/hg19/ensembl_hg19.events.ioe -e iso_tpm_formatted.txt -o HSCR_events
# hg38
python ~/softwares/SUPPA/suppa.py psiPerEvent -i ~/references/SUPPA2/ref/GRCh38/gencode.v37.all.events.ioe -e iso_tpm_formatted.txt -o HSCR_events
# error - skip some AS events
ERROR:psiCalculator:transcript ENST00000484026.6_PAR_Y not found in the "expression file".
ERROR:psiCalculator:PSI not calculated for event ENSG00000169100.14_PAR_Y;SE:chrY:1389727-1390192:1390293-1391899:-.
# PKM MX
ENSG00000067225;MX:chr15:72492996-72494795:72494961-72499069:72492996-72495363:72495529-72499069:-
# qPCR testing samples
IMR90.list 17C8.list
# PKM result
event X17C8.list00_output X17C8.list01_output X17C8.list02_output X17C8.list03_output X17C8.list04_output IMR90.list00_output IMR90.list01_output IMR90.list02_output IMR90.list03_output IMR90.list04_output
ENSG00000067225.19;MX:chr15:72200655-72202454:72202620-72206728:72200655-72203022:72203188-72206728:- 0.9220111807188586 0.9218209128593989 0.9369499000184589 0.9371596717647238 0.9437735074216145 0.9358327299524112 0.9162359515407531 0.9017577693324474 0.9236993945732118 0.9051320273934063
最好用最新的hg38的注释(genecode v37),否则AS数据会非常不全。
suppa这个工具还是不错的,AS的种类丰富,结果也比较靠谱。
发现一个课题组,已经在AS分析上做了大量的工作,值得学习。
- VAST-TOOLS - GitHub
- VastDB - An atlas of alternative splicing profiles in vertebrate cell and tissue types
- Matt - A Unix toolkit for analyzing genomic sequences with focus on down-stream analysis of alternative splicing events
通过可视化工具了解的信息【不会可视化,你就永远都看不懂结果】:
junction:从一个exon的结束到另一个exon的起点,这就是一个junction,俗称跳跃点、接合点,因为这两个点注定要连在一起。
AS event:一个可变剪切的时间,如何定义呢,一个exon,以及三个junction即可定义,要满足等式相等原则,即一个exon的exclusion。
核心需求:
- 单细胞的整体PSI分析【whole genome】
- 单细胞、bulk的特定的exon的PSI value【outrigger】
- target gene exon PSI value
- 单细胞、bulk的特定的exon的表达值
重点看:
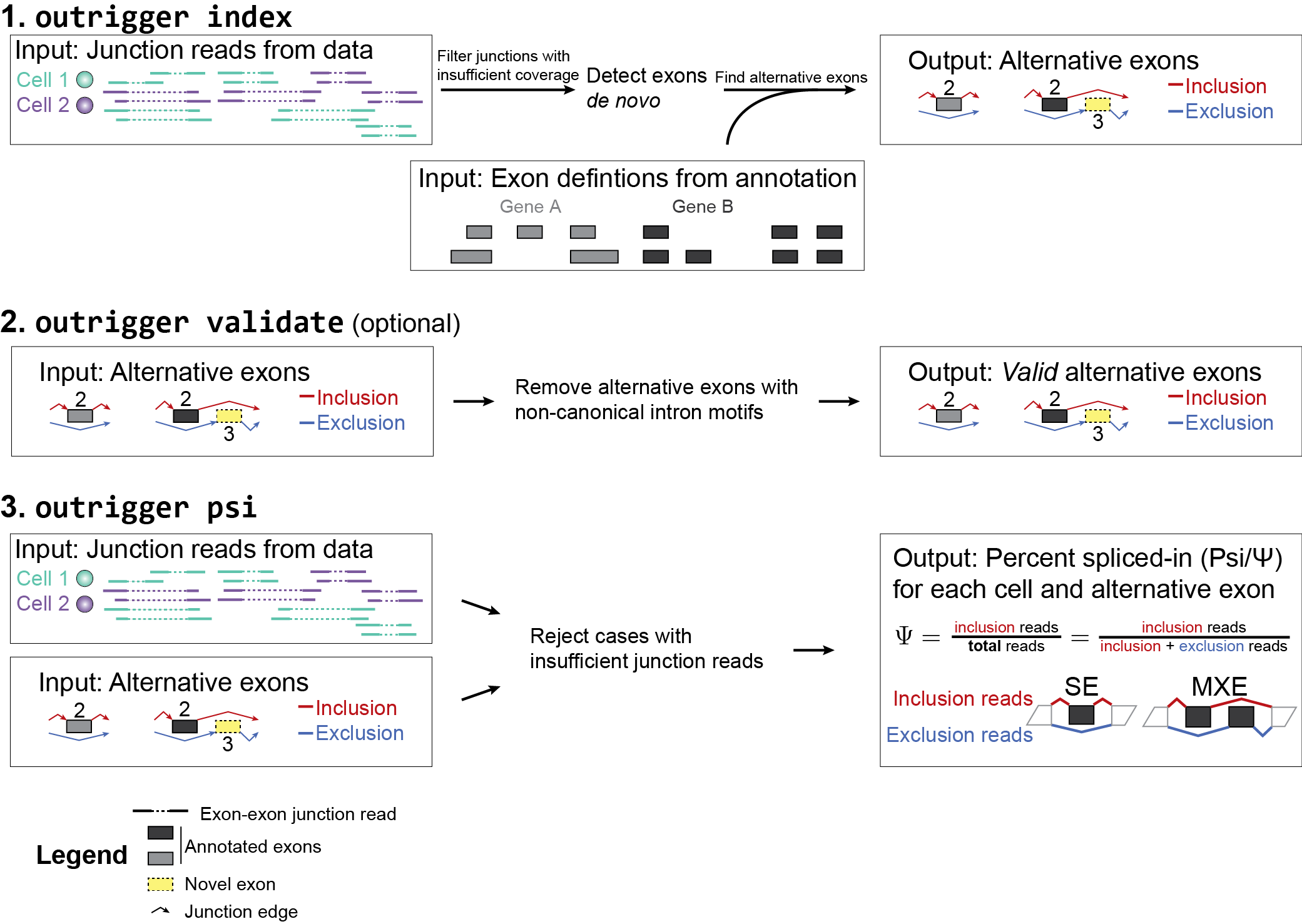
index: Build a de novo splicing annotation index of events custom to your data
Google search:get PSI value for one exon【PSI值没那么好算,最好把它底层的逻辑搞懂】
PSI,非常简单明确,就是inclusion reads / (inclusion reads + exclusion reads)
inclusion reads比较明确,按single end来看,凡事比对到目标exon的reads都算作inclusion reads;
exclusion reads则没那么直接,不是所有的减去inclusion reads,而是跳过目标exon的reads,其他不相干的reads都不算;
https://github.com/JY-Zhou/FreePSI
http://projects.insilico.us.com/TCGASpliceSeq/faq.jsp
根据bulk RNA-seq或者single-cell RNA-seq来找isoform的类型。【难点:二代NGS的reads长度较短,可能无法成功组装出full-length的isoform。】
我们只需要关注junction read,基数即可。
junction read:在成熟mRNA中,测序的reads如果同时跨越了两个exon,则为junction read。
如何提取出junction read?【不靠谱,用outrigger,里面有个reads.csv文件】
/home/lizhixin/softwares/regtools/build/regtools junctions extract -r chr19:797075-812327 -s 0 UE.bam
自己写一个程序吧,用bedtools coverage这个功能。
bedtools coverage -a regions.bed -b reads.bam
spliceGraph
核心就是count reads,同时比对到两个点上。【这个不难,但是并不能说明问题】
samtools view -u -@ 2 UE.bam chr19:803636-803636 | less -S
必须鉴定出拼接两个exon的read,这样的才是有意义的证据,单纯的连接的数据并不实用【中间可能插入了很多个exon,连接≠拼接】。
cat gencode.v27.annotation.gtf | grep ""PKM"" | awk '{if($3=="exon")print $0}' > PKM.txt
samtools view -b -@ 2 23c9.bam chr19:797075-812327 > PTBP1.23c9.bam
参考:
Question: Count Junctions In A Sam/Bam File
Question: How Many Reads In A Bam File Are Aligned To a Specific Region