连锁不平衡 linkage disequilibrium (LD) 又称为等位基因关联(allelic association)
广泛的遗传关联研究依赖genetic marker和disease locus的连锁不平衡(linkage disequilibrium,LD)
http://www.voidcn.com/article/p-aifzvzzo-bpz.html
连锁不平衡(linkage disequilibrium,LD)是指在某一个群体中,不同座位上两个基因同时遗传的频率明显高于预期的随机频率现象。
We have developed an approach,LD Score regression, that quantifies the contribution of each by examining the relationship between test statistics and linkage disequilibrium (LD).
LD四大功能:estimating LD Scores, h2 and partitioned h2, genetic correlation, the LD Score regression intercept。
计算LD分数、性状的遗传度、性状间的遗传相关性及遗传协方差,分割遗传度,细胞类型特异性分析等
表示LD的两个度量指标,D'和r2来表示LD的程度。
当D‘=1,表示连锁完全不平衡,没有重组;
当D‘=0,表示连锁完全平衡,随机组合;
当r2=1,表示连锁完全不平衡,没有重组;
当r2=0,表示连锁完全平衡,随机组合。
r2包括了重组和突变,而D’只包括重组史。与D'相比,在同样长度的染色体范围内,r2往往更低,这个特性能够帮助我们找到更精度的基因定位。另外,r2和D'相比,受样本量和等位基因频率的影响较小。D’能更准确地估测重组差异,但样本较小时低频率4种等位基因组合的可能性大大减小,因此D’不适合小样本研究。LD作图中通常采用r2来表示群体的LD水平
if the sample size is increased by a factor of 1/r2, where r2 is the commonly used measure of pairwise LD.
我们一般用D,D'和r2来表示LD的程度。
(Delta)D是LD的基本单位,度量观察到的单倍型频率与平衡状态下期望频率的偏差,算法如下:
D=P(AB)-P(A)*P(B); (PAB is the expectant frequency of AB haplotype, and P(A)*P(B) is the actual frequency)
P(AB)表示实际观察到的AB频率,P(A)*P(B)表示AB频率的期望值。(如果发生连锁不平衡,实际观测到的AB频率肯定不等于AB频率的期望值)
r2=D*D/(P(A)P(a)P(B)P(b))。 注:小写的r
当r2=1,表示连锁完全不平衡,没有重组
当r2=0,表示连锁完全平衡,随机组合
r2变化于0到1之间,反映两个位点之间的“correlation coefficient”;r2=1,提示两位点将产生完全相同的信息(所以,选择tagSNP时会参考r2);r2=0,提示遗传平衡;同样r2在0到1之间变化与可反映连锁不平衡的程度。
r2=0.001,如果某一个位点的一个基因频率特别低的话,则依据r2值,判断两位点接近连锁平衡。
同时,r2=1有更严格的解释:两个位点的等位基因有相同的频率,并且一个位点某个等位基因的出现完全预示着另外一个位点相应等位基因的出现,这时候两个位点组成的四种可能的单倍型仅表现为两种。与D'相比,r2在连锁不平衡中更加有用。
http://yangli.name/2016/05/10/20160510snpld/
*D’= Standardized D; r=pearson coefficient of correlation
- D’=D/Dmax (Dmax=min(PAPb,PaPB)
- r2 =D/PAPBPa*Pb
http://blog.sciencenet.cn/blog-797870-659792.html
disequilibrium [ˌdɪsˌiːkwɪˈlɪbriəm,] 不平衡
correlation coefficient : 相关系数
R平方 :大写的R 用于回归评价指标,R2方法是将预测值跟只使用均值的情况下相比,看能好多少。其区间通常在(0,1)之间。0表示还不如什么都不预测,直接取均值的情况,而1表示所有预测跟真实结果完美匹配的情况。1-(残差平方和/总体平方和)用来反映数据和模型的拟合程度。
https://www.jianshu.com/p/a36bd4145ef7
D衰减图就是利用曲线图来呈现基因组上分子标记间的平均LD系数随着标记间距离增加而降低的过程。大概的计算原理就是先统计基因组上两两标记间的LD系数大小,再按照标记间的距离对LD系数进行分类,最终可以计算出一定距离的分子标记间的平均LD系数大小。如图3是黄瓜重测序文章中统计各个亚群体的LD衰减速度的图形。横坐标是物理距离(kb),纵坐标是LD系数(r^2)。
一般而言,LD系数大于0.8就是强相关。如果LD系数小于0.1,则可以认为没有相关性。如果LD衰减到0.1这么大的区间内都没有标记覆盖的话,即使这个区间有一个效应很强的功能突变,也是检测不到关联信号的。所以,通常可以通过比较LD衰减(到0.1)距离和标记间的平均距离,来判断标记是否对全基因组有足够的覆盖度。(GWAS标记量=基因组大小/LD衰减距离)
Haploview 生成连锁图
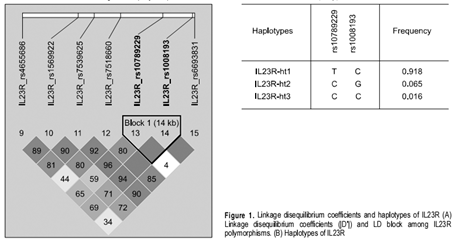
小方框里的数据表明r2或D'的数值,颜色越深,表明MARKER之间LD的强度越大.BLOCK是软件自动生成的,一般表明是完全连锁的,14KB表明两MARKER之间的距离
Haploview 输入两个文件,ped基因分型结果和info包含SNP位点ID和位置信息。
Haploview生成的LD plot,每个格子代表了两个SNP位点之间的LD分析结果,颜色从白色到红色,代表连锁程度从低到高。方框中的数值为D’值乘以100.相互之间高度连锁的SNP位点构成了haplotype block,如下图1-8构成block1,长度为84kb。参考https://blog.csdn.net/weixin_43569478/article/details/108079154
