在使用FastQC之后,如果我们发现了一些问题(序列质量不高),那么我们该使用什么样的工具,去解决这些问题呢?
fastx Toolkit是包含处理fastq/fasta文件的一系列的工具,它是基于java开发的,我们高通量测序最常用到的是使用这个软件进行reads的裁剪(trim)
FASTQ-to-FASTA
说明:这个命令主要是用来转换FASTA格式与FASTQ格式
fastq_to_fasta [-h] [-r] [-n] [-v] [-z] [-i] [-o]
[-h] = 获得帮助信息
[-r] = 使用序号去代替fastq文件中原来的reads名
[-n] = 如果fastq中有N,保留(默认是有N的序列删除)
[-v] = 报告reads的总数
[-z] = 调用GZip软件,输出的文件自动经过压缩
[-i] = 输入文件,可以是fastq/fasta格式
[-o] =输出路径,如果不设置会直接输出到屏幕
FASTX Statistics
说明:主要统计一下序列的基本信息,如GC含量什么的,很少用,基本使用FastQC代替
fastx_quality_stats [-h] [-i INFILE] [-o OUTFILE]
[-h] = 获得帮助信息
[-i] = FASTA/Q格式的输入文件
[-o] = 输出路径,如果不设置会直接输出到屏幕
FASTA/Q Clipper
说明:主要是进行reads的过滤和adapter的裁剪
fastx_clipper [-h] [-a ADAPTER] [-i INFILE] [-o OUTFILE]
[-h] = 获得帮助信息
[-a ADAPTER] = Adapter序列信息,默认的是CCTTAAGG
[-l N] = 如果1条reads小于N就抛弃,默认5
[-d N] = 保留adapter并保留后面的Nbp,如果设置-d 0等于没有用这个参数
[-c] = 只保留包含adapter的序列
[-C] = 只保留不包含adapter的序列
[-k] = 报告adater的序列信息
[-n] = 如果reads中有N,保留reads(默认是有N的序列删除)
[-v] = 报告序列总数
[-z] = 调用GZip软件,输出的文件自动经过压缩
[-D]= Debug output
FASTA/Q Trimmer
说明:这个是我最常用的工具,可以快速切序列
fastx_trimmer [-h] [-f N] [-l N] [-z] [-v] [-i INFILE] [-o OUTFILE]
[-h] = 获得帮助信息
[-f N] = 序列中从第几个碱基开始保留,默认是1
[-l N] = 序列最后保留到多少个碱基,默认是整条序列全部保留
[-z] = 调用GZip软件,输出的文件自动经过压缩
cutadapt软件
这个cutadapt软件是最常用的去adapter的工具。它是基于Python编写的一个Python包
一些最基本的用法
# cutadapt的功能特别强大,相对应的参数真的特别多,有几十个参数,我们平时只会用到很少的几个,我在这里为大家介绍一下。
# 最基本的形式,可以去掉3‘端的adapter序列
cutadapt -a AACCGGTT -o output.fastq input.fastq
# 可以直接输入或者输出压缩文件,不需要修改参数,输出文件的后面加上.gz
cutadapt -a AACCGGTT -o output.fastq.gz input.fastq.gz
# 假如去掉3‘端的adapter AAAAAAA 和5’端的adapter TTTTTTT
cutadapt -a AAAAAAA -g TTTTTTT -o output.fastq input.fastq
# cutadapt也可以用来进行reads的cut,去掉最前面的5bp
cutadapt -u 5 -o trimmed.fastq input_reads.fastq
# 进行reads测序质量的过滤
# cutadapt软件可以使用-q参数进行reads质量的过滤。基本原理就是,一般reads头和尾会因为测序仪状态或者是反应时间的问题造成测序质量差,比较粗略的一个过滤办法就是-q进行过滤。需要特别说明的是,这里的-q对应的数字和phred值是不一样的,它是软件根据一定的算法计算出来的
# 3‘端进行一个简单的过滤,--quality-base=33是指序列使用的是phred33计分系统
cutadapt -q 10 --quality-base=33 -o output.fastq input.fastq
# 3‘端 5’端都进行过滤,3'的阈值是10,5‘的阈值是15
cutadapt -q 10,15 --quality-base=33 -o output.fastq input.fastq
Reads 长度的过滤
[--minimum-length N or -m N]
# 当序列长度小于N的时候,reads扔掉
[--too-short-output FILE]
# 上面参数获得的这些序列不是直接扔掉,而是输出到一个文件中
[--maximum-length N or -M N]
# 当序列长度大于N的时候,reads扔掉
[--too-long-output FILE]
# 上面参数获得的这些序列不是直接扔掉,而是输出到一个文件中
Paired-Reads的裁剪(trim)
# 现在很多的测序都是双端测序,那么从测序原理上来说,一对reads来自于1簇反应,所以一起进行adapter的trim可能效果更好。cutadapt自然也提供了这样的功能
cutadapt -a ADAPTER_FWD -A ADAPTER_REV -o out.1.fastq -p out.2.fastq reads.1.fastq reads.2.fastq
# -a是第1个文件的adapter序列
# -A是第2个文件的adapter序列
# -o是第1个输出文件
# -p是第2个输出文件
1个例子
其实在实际中,我们从公司拿到的数据大多数已经进行过cutadapt,我们其实更关注的是reads的trim
我们首先要用fastqc对test1.fastq与test2.fastq进行一下质量评估,评估的主要结果如下:
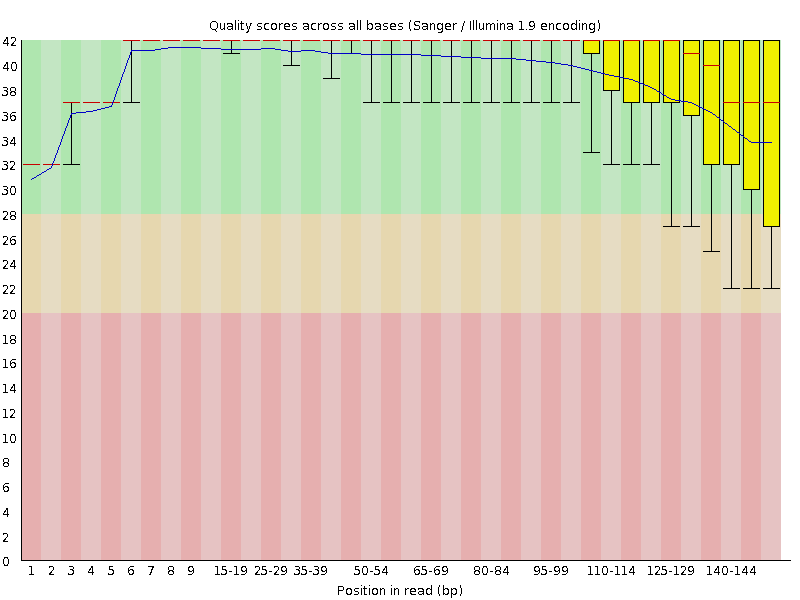
read1的测序质量图
read2的测序质量图
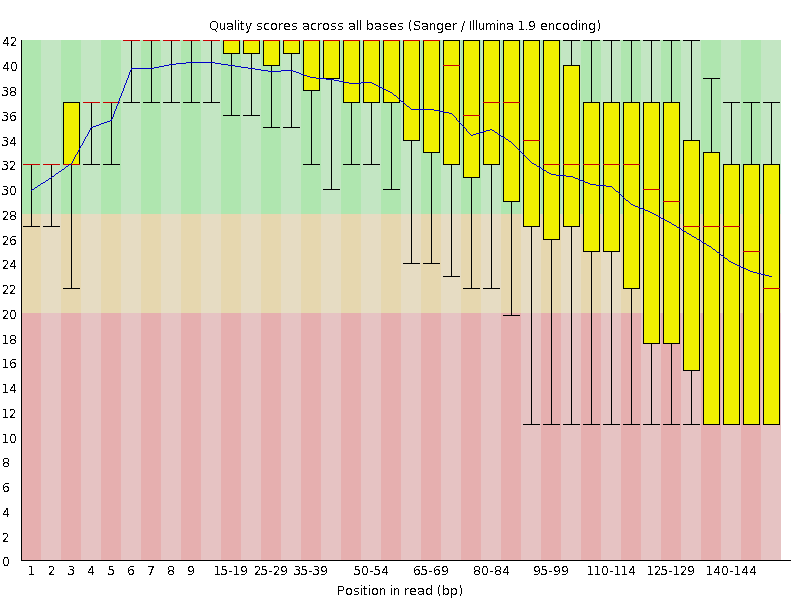
我们从上面两张图可以明显看出,read1的测序质量明显好于read2,一般我们确定要trim多少bp的时候都是按phred20这个标准进行评估。比如,对于我们测试数据,read1就不需要trim,read2需要保留1-85bp。对应的fastx_trimmer的命令如下:
fastx_trimmer -i test_data_2.fastq -o test_data_2_trim.fastq -f 1 -l 85
[-f N] = 序列中从第几个碱基开始保留,默认是1
[-l N] = 序列最后保留到多少个碱基,默认是整条序列全部保留