数据分析与解读
1. Data Cleaning
从原始数据(Raw Data)到干净数据(Clean Data)的过程,有人翻译成“数据清洗”,实在叫不习惯
Illumina测序仪下机的数据通常为Bcl格式,是将同一个测序通道(Lane)所有样品的数据混杂在一起的,所以公司一般不会提供Bcl文件。测序公司使用Illumina官方出品的Bcl2FastQ软件,根据Index序列分割转换成每个样品的FastQ文件,打开长这样:

原始数据没法直接分析,是因为部分reads测序质量较低,可能会误导后续结果,因此需要对低质量碱基太多或N(未能识别的碱基)太多的reads进行去除;此外,部分测序文库的插入片段太短,导致测到两侧的接头序列,这些序列接头也需要从reads中去除。最后,我们也会对清洗前后的Raw Data和Clean Data进行评估,评估内容包括碱基质量、序列长度、碱基比例、GC含量、重复序列、Kmers等
#常用软件#
我以前都是用cutadapt + FASTX-Toolkit的组合,直到同事们给我推荐了Trim Galore,质量评估使用FastQC
1.5 #可选步骤# 核糖体RNA(rRNA)去除
2. 比对
如果不是很急或者只想知道已知转录本表达量,个人建议使用基因组比对的方法进行分析,理由如下:
① 转录组比对需要准确的已知转录本的序列,对于来自未知转录本(比如一些未被数据库收录的lncRNA)或序列不准确的reads无法正确比对;
② 与上一条类似,转录组比对不能对转录本的可变剪接进行分析,数据库中未收录的剪接位点会被直接丢弃;
③ 由于同一个基因存在不同的转录本,因此很多reads可以同时完美比对到多个转录本,reads的比对评分会偏低,可能被后续计算表达量的软件舍弃,影响后续分析;
④ 由于与DNA测序使用的参考序列不同,因此不利于RNA和DNA数据的整合分析。
此外,值得注意的是,RNA测序并不能直接使用DNA测序常用的BWA、Bowtie等比对软件,这是由于真核生物内含子的存在,导致测到的reads并不与基因组序列完全一致,因此需要使用Tophat/HISAT/STAR等专门为RNA测序设计的软件进行比对
比对结果会展示为BAM/SAM文件,其中BAM格式是SAM格式的二进制版本
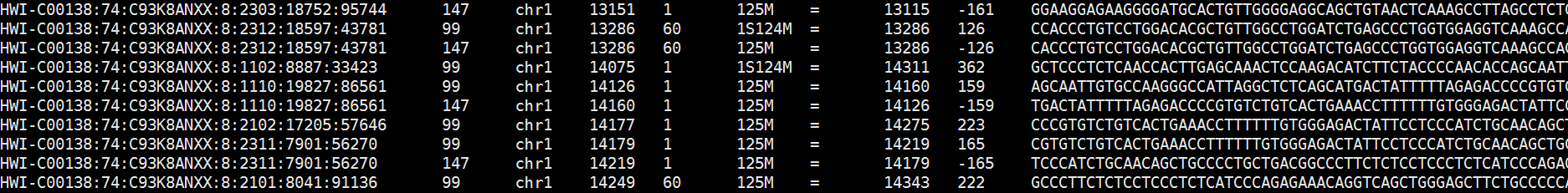
BAM文件中每行代表一条reads的比对信息,其中第一列是read的ID,第二列为FLAG(包括是否双端比对,比对位点是否唯一等信息),第三列为比对的染色体,第四列为比对的起始位置,第六列为CIGAR值,代表比对的具体方式(例60M2D80M代表60个碱基完美匹配+2个碱基缺失+80个碱基完美匹配)等等
#常用软件#
基因组比对:
Tophat2:可以说是最被公认的RNA测序比对软件(实际上是在DNA比对软件Bowtie的基础上做了一个壳),相信很多做RNA测序的同学都是看着Tophat发表在Nature Protocol上的步骤一步步入门RNA测序的;
HISAT2:Tophat2的非正式升级版本(因为据说还会有Tophat3),在Tophat的算法基础了上做了大量的改进,而且克服了Tophat最大的缺点——速度慢,Nature Protocol上同样发表了操作流程;
STAR:ENCODE计划御用比对软件,权威程度可以与Tophat平起平坐,并且比对速度极快;
MapSplice:TCGA使用的比对软件,我自己没用过;
RSEM:RSEM更像一个软件包而不是一个比对软件,能够提供从比对到计算差异表达的所有步骤,由于不需要自己写代码串联不同软件生成的数据格式,因此用起来比较省时省力,值得注意的是,TCGA使用MapSplice比对后再用RSEM计算表达量,并没有直接只用RSEM原装的Bowtie的比对结果。
转录组比对:这类型的软件我用的不多,最近尝试过Nature Methods上面发表的Salmon,能从Clean reads直接算到表达量,优点是,快,非常快。然而这个软件连BAM文件都没生成,虽然只是定量的话BAM文件的确没什么用就是了…