Chen Sun, Zhiqiang Hu, Tianqing Zheng, Kuangchen Lu, Yue Zhao, Wensheng Wang, Jianxin Shi, Chunchao Wang, Jinyuan Lu, Dabing Zhang, Zhikang Li, Chaochun Wei, RPAN: rice pan-genome browser for ∼3000 rice genomes, Nucleic Acids Research, Volume 45, Issue 2, January 2017, Pages 597–605, https://doi.org/10.1093/nar/gkw958
“Pan-genome”是指一个clade或品种中的所有个体都包含的一组基因集,其提供一个新的维度来进行研究,即通过基因在基因组上的存在或缺失来评价。在该研究前不久,the 3000 Rice Genome Project (3K RGP)以平均深度14.3x测序了超过3000个水稻基因组。本研究作者筛选出了水稻的pan-genome,并开发了基因组浏览器Rice Pan-genome Browser (RPAN)来研究从3K RGP得到的pan-genome。
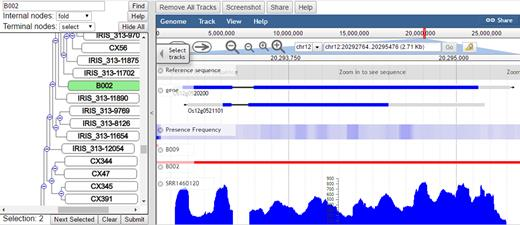
RPAN可以查询3010个水稻accession的基本信息包括基因组序列、基因注释、PAV(基因出现/缺失变异)信息和基因表达信息,在参考基因组中缺失的至少12000个novel基因被包含在内。RPAN还提供了多种搜索与可视化功能。
为了构建pan-genome,作者将每个accession的原始测序数据先用SOAPdenovo version r240进行组装,随后通过Mummer package version 3.23将长度超过500bp的contig比对到IRGSP基因组上。对于未比对的contig,采用CD-HIT删除了冗余的序列(识别阈值为90%),随后各种污染物(various contaminants)被使用NCBI-blast通过NT数据库去除,接着通过NCBI-blast执行all-vs-all比对来确保没有冗余序列。剩余的contig组成了非冗余的novel序列数据集。所有的经过筛选、非冗余的未比对的序列被归入12个组,这些组是根据SNP分析事先定义出的population structure。所有分在一个组的contig用100个连续的“N”相连接。最后,IRGSP基因组(373Mbps)与这些novel序列(268Mbps)合并到了一起,作为水稻的参考pan-genome。
pan-genome中,未比对上的部分序列的蛋白编码基因采用MARKER-P和IRGSP-1.0基因组进行预测。
为了进行基因存在/缺失判断,作者首先将水稻accession所有的原始读段被使用BWA version 0.7.10的“bwa mem”来map到pan-genome序列上。通过samtools version 0.1.19,拥有编码区覆盖率大于0.95、整个基因覆盖大于0.85的基因被认为在该accession上存在。
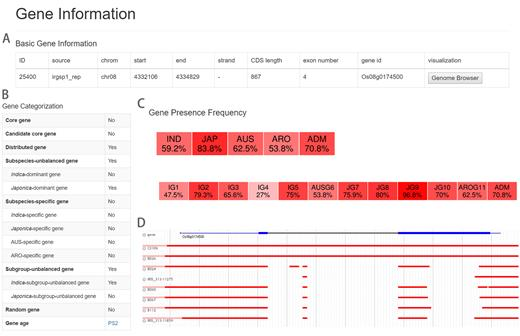
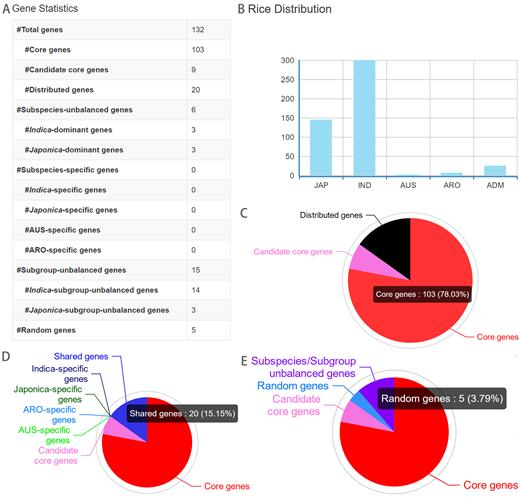
基因被按照其在所有accession上的存在/缺失情况归档。作者对于基因的存在/缺失检测中仅采用了453个高质量(深度大于20x,比对深度大于15x)accession的数据。作者对在所有453个水稻accession中存在的基因(核心基因)和其他基因做了分布图。为了减少错误发现率,作者仅保留了缺失样本大于1%的,而其他的(被作者称为核心基因的候选基因)则被从distributed gene set中移除。Distributed gene被进一步按照subspecies进行分组。并在进一步区分中提出了“random gene”的概念。Random基因在亚种间或亚种内部的亚组间不呈现出频率上的显著差异。
接着,作者还准备了RNA-seq数据作为表达数据。
在数据库的构建上,作者同时采用了基于JBrowse的框架和基于HTML5、SVG和JavaScript。后者被用于绘制进化树。