缺点:需要联网,经常出错,不是操作问题而是因为网络问题
安装
if("seqinr" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("seqinr")}
suppressMessages(library(seqinr))
ls('package:seqinr')
###Retrieving a sequence and write into FASTA file###
1) 选择要去fetch序列的数据库(这里已genebank为例)
choosebank() #查看有哪些数据库
choosebank('genbank')

2)一旦选择好了数据库,用query信息进行收索
BRCA1<- query("BRCA1", "SP=Homo sapiens AND K=BRCA1")
3)查看query返回的对象所有属性
attributes(BRCA1) mynames <- getName(BRCA1) #查看所有搜索到的名称 length(mynames) #查看共检索到多少,写入到文档的时候可以用来用来检查 209
![]()
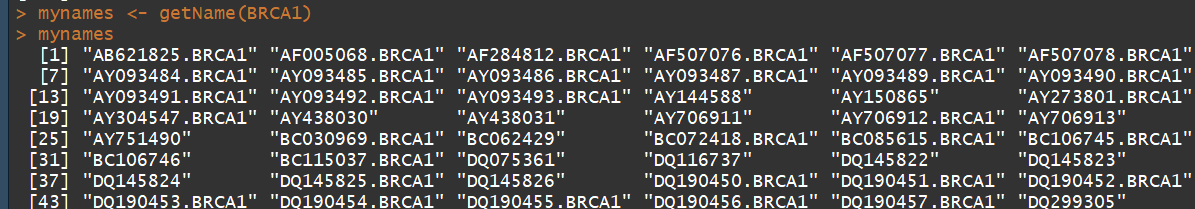
4)查看所有收索到的序列所包含的属性
BARC1$req

5) 获取所有检索到的序列,并以fasta格式写入到文件中
all_myseqs <- getSequence(BRCA1) #所有收索到的序列 write.fasta(all_myseqs, mynames, file.out = "MyBRCA.fasta") #将所有收索到的序列写入fasta格式文件。
###2、Getting the detail of a sequence composition###
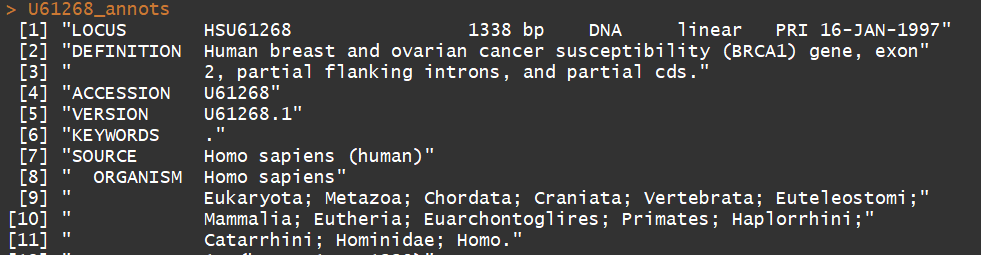
6)提取上述特定的序列(第一条序列),及注释信息
myseq <- getSequence(BRCA1$req[[1]])
annots <- getAnnot(BRCA1$req[[1]]) myseq

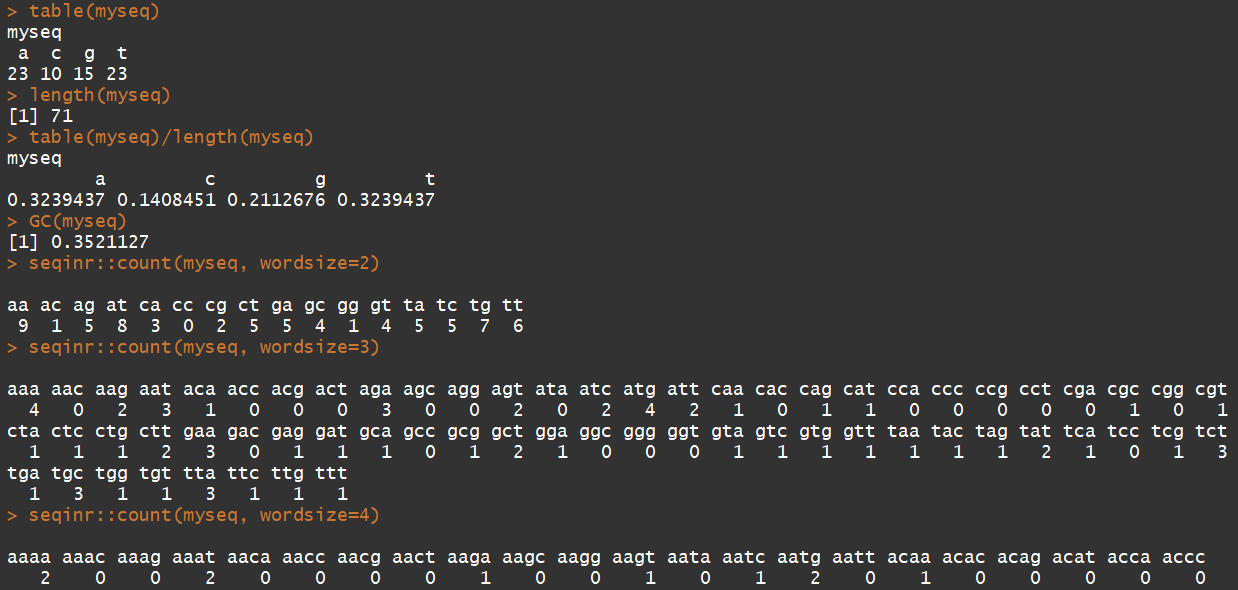
7)统计上述序列中各个碱基的含量
table(myseq) #统计该序列中碱基使用情况 length(myseq) #计算序列长度 table(myseq)/length(myseq) #统计碱基百分比 GC(myseq) #计算GC含量 seqinr::count(myseq, wordsize=2) #以2个碱基出现频率 seqinr::count(myseq, wordsize=3) #以三个碱基出现频率 seqinr::count(myseq, wordsize=4) #以四个碱基出现频率 seqinr::count(myseq, wordsize=5) #以5个碱基出现频率
6)关闭接口,防止打开多个接口
closebank()
除了上述之外,如果你知道ID号码也可以根据数据库AC属性 (AC attribute)提取搜索到的特定序列
U61268<-query("BRCA1", "SP=Homo sapiens AND AC=U61268") #也可以根据特定的ID进行搜索
attributes(U61268)
U61268$req
U61268_seq <- getSequence(U61268$req[[1]])
U61268_annots <- getAnnot(U61268$req[[1]])