1. 起因
之前的代码(单细胞分析实录(17): 非负矩阵分解(NMF)代码演示)没有涉及到python语法,只有4个python命令行,就跟Linux下面的ls grep一样的。然鹅,有几个小伙伴不会命令行,所以我决定再改写一下,把命令行都放到R下面运行。
2. 尝试
2.1 一开始,我的想法是教大家在R里面调用python,需要提前下载好anaconda和一些python包
然而想了想在Windows上安装python包可能对大家不是很友好,有些包很难装,我之前也弄了很久。考虑到这次更新是针对桌面版Rstudio用户,故没有采用。
2.2 最终,我采用的方案是,使用Rstudio Server,也就是网页版Rstudio
这样做有几个好处:
- 直接和云服务器连接,服务器下载python包和R包都很容易(云服务器刚买,下血本)
- 我提前配置好运行环境,用户只需上传数据,分析数据,下载数据即可。
代码方面也更加简化:
- 我尽量减少了人工处理的时间,主要分析代码只有两行
如果你之前在我这儿拿过代码,可以直接找我要更新的代码。此外,如果因为之前的代码涉及命令行,你操作起来有困难,可以找我开Rstudio Server的账户 (高端玩家就别了,服务器配置比较低,就够几个人用的那种)。
3. 注意
- 我会提前安装可能用到的R包,所以不用重复安装,直接library就可以
- 请大家及时下载结果文件,以免丢失;也请大家在做完分析后,删除表达数据,服务器存储空间不是很大
- 每个账号只保留半个月时间,若想再次使用,可以联系我再开一个账号
- 有任何问题可以微信或者邮箱问我
接下来简单介绍一下,使用方法
登录
打开我给你的链接,输入用户名和密码即可登录
之后就可以看见Rstudio的界面了

然后确保你的家目录下面有图中框出来的几个文件,并点击进入count_data文件夹
上传数据
点击upload上传数据

运行代码
主要是3.R中的step1和step2两个函数

library(reticulate)
use_condaenv(condaenv = "cnmf_env", required = T,conda = "/home/hsy/miniconda3/bin/conda")
py_config() #如果显示cnmf_env环境里面的python就OK
source("1.R")
step1(dir_input = "count_data",dir_output = "res1",k=3:5,iteration = 50) #这里为了演示方便,取值都比较小
source("2.R")
step2(dir_input = "res1",dir_output = "res2",dir_count = "count_data",usage_filter = 0.03,top_gene = 30,cor_min = 0,cor_max = 0.6)
查看结果
step2之后,会在res2文件夹中生成结果文件
sampleID_program.usage.norm.txt和sampleID_program.Zscore.txt
是NMF分解表达矩阵得到的两个矩阵
program_topngene.txt
这是所有program的前几十个基因,一般会放到文件附表
program_pearson_cor.complete.heatmap.pdf
program之间的相关性热图
cor_heatmap_data.txt
用来画上图的数据
program_topngene_enrichment.xlsx
program_topngene_enrichment_order.csv
这两个都是对program前几十个基因的富集分析结果,这两个文件可以用来辅助我们理解program,其中第二个文件和相关性热图的顺序一致,看起来更方便
sampleID_program_gene.heatmap.pdf
用来验证在这个样本中,program找得对不对,其实就是看program的表达,一般看program的前几十个基因
sampleID_data_heatmap.txt
用来画上面那个热图的数据
program之间的相关性热图

某个样本中program的表达

下载结果
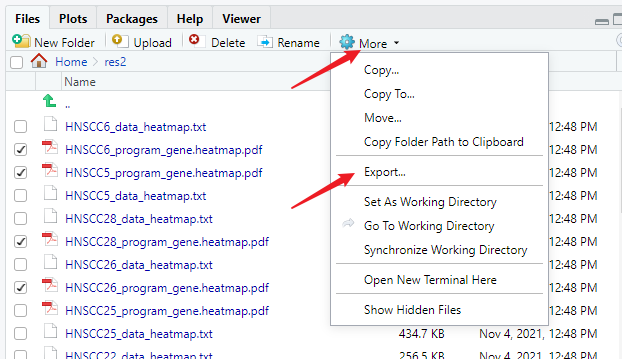
选中你想导出的文件,点击more,再点击Export就可以了
至此,公众号仅有的两篇付费教程都已更新完毕~
因水平有限,有错误的地方,欢迎批评指正!