Update 7.1
计算部分
计算部分通过另一名同学看慕课对广义线性模型的学习,基于现有软件的计算部分实现了一份基于intel mkl的关联分析程序。上面我们解析完成后得到了包含bgen和sample的混合数据,和同组的同学商量好对接的接口之后直接调用计算部分。
首先是数据的组合转化过程:

这里读完所有的变异并转化成snps,即直到count == number_of_variants的时候停下,然后把上面的混合数据和这里的变异信息一起传给计算部分进行关联分析,现在调试阶段就规定每chunk个变异就计算一次。
计算过程:

为了获取优化的热点,我们对解析和计算部分分别计时。到这一步的运行结果:
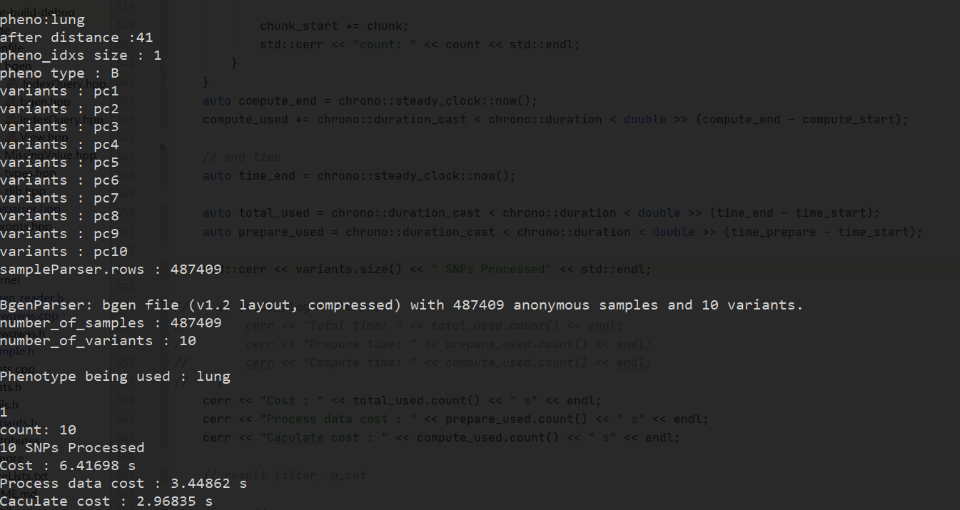
可以看到数据解析部分占用的时间略微的高于计算部分。
并行优化
这一块是整个项目的重点和难点,也是我们的gwas软件相比现有软件的创新之处。
首先,我们用C语言实现的初始版本应该就比现有的R语言的软件要快不少:
| 版本 | 处理1000个snp花费时间/秒 |
|---|---|
| SNPTest | >18000 |
| Baseline | 244.24 |
首先,单线程下的常数优化。我们设置的chunk相当于缓冲区的大小,即每读chunk个snp就进行计算,chunk太大内存放不下,太小的话频繁的在读入和计算之间切换会导致效果不好,我们用比较原始的方法,枚举chunk大小来比较时间,从而确定下chunk的值:
snpNumber=1000
| chunk | cost/秒 |
|---|---|
| 1 | 208.77 |
| 2 | 181.51 |
| 5 | 151.82 |
| 10 | 137.95 |
| 20 | 212.92 |
| 50 | 224.99 |
| 100 | 244.24 |
这里chunk最大就设置到100,如果再大的话可能内存需求超过4G,最终确定chunk的大小为10。
经过chunk的优化,现在的性能如下:
| 版本 | 处理1000个snp花费时间/秒 |
|---|---|
| SNPTest | >18000 |
| Baseline | 244.24 |
| Chunk | 138.95 |
下面考虑多线程并行。
由于最最开始就决定计算部分是用intel mkl优化的,mkl自带了并行,是在最内层调用相关的函数的时候并行的,我们简单看一下效果:(处理1000个snp chunk=10)
设置线程数:export MKL_NUM_THREADS=1
| 线程数 | 运行时间(总时间=处理数据+准备数据+计算) |
|---|---|
| 1 | 137.95=2.32+21.56+114.07 |
| 2 | 130.95=2.25+23.58+105.12 |
| 4 | 109.10=2.34+22.34+84.42 |
| 8 | 101.88=2.25+24.28+75.35 |
| 16 | 104.55=2.33+22.55+79.67 |
可以看到,加速比很低,16线程的时候甚至出现了性能下降。经过分析之后,原因应该是最内层的intel相关函数并不是热点,而且16线程的时候,在线程调配上花了更多的时间。
下面我们抛弃mkl自带的并行策略,自己手动实现并行。
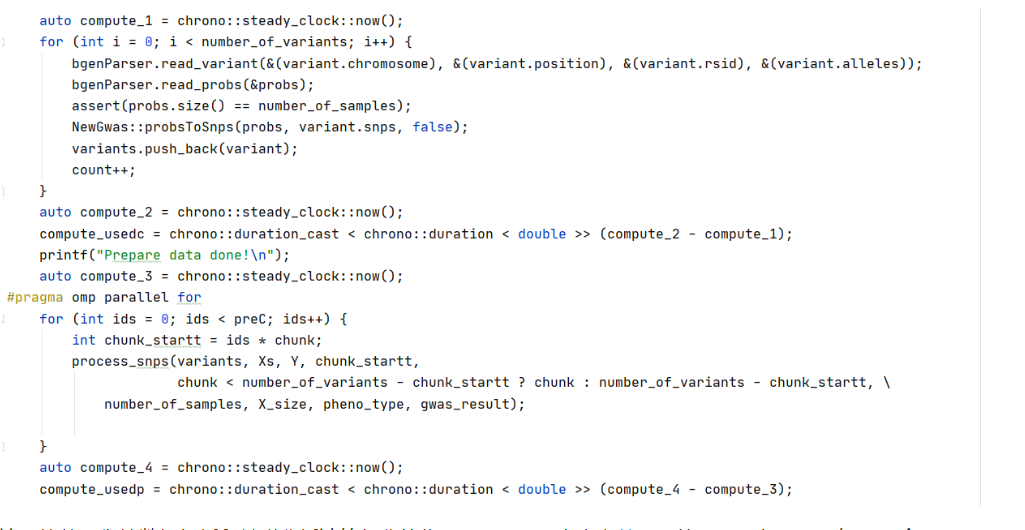
这一块就是衔接数据解析和关联分析计算部分的代码了,chunk大小之前设置的是10,如果snp有1000个,那preC=100,所以并行加在这个100大小的循环上是比较合适的,我们直接利用openMP,在这个循环上进行并行。
设置线程数:export OMP_NUM_THREADS=1
然后具体的运行时间如下:(处理1000个snp chunk=10)
| 线程数 | 运行时间(总时间=处理数据+准备数据+计算) |
|---|---|
| 1 | 137.95=2.32+21.56+114.07 |
| 2 | 101.66=2.41+23.58+75.67 |
| 4 | 64.53=2.30+21.52+40.71 |
| 8 | 58.98=2.47+22.84+33.67 |
| 16 | 56.18=2.37+23.92+29.89 |
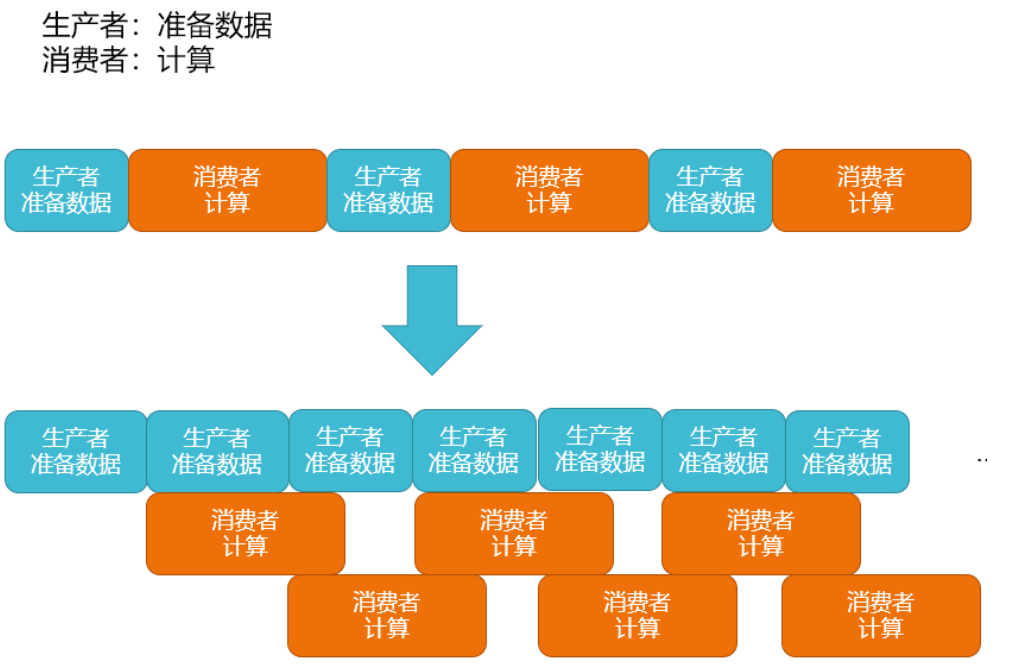
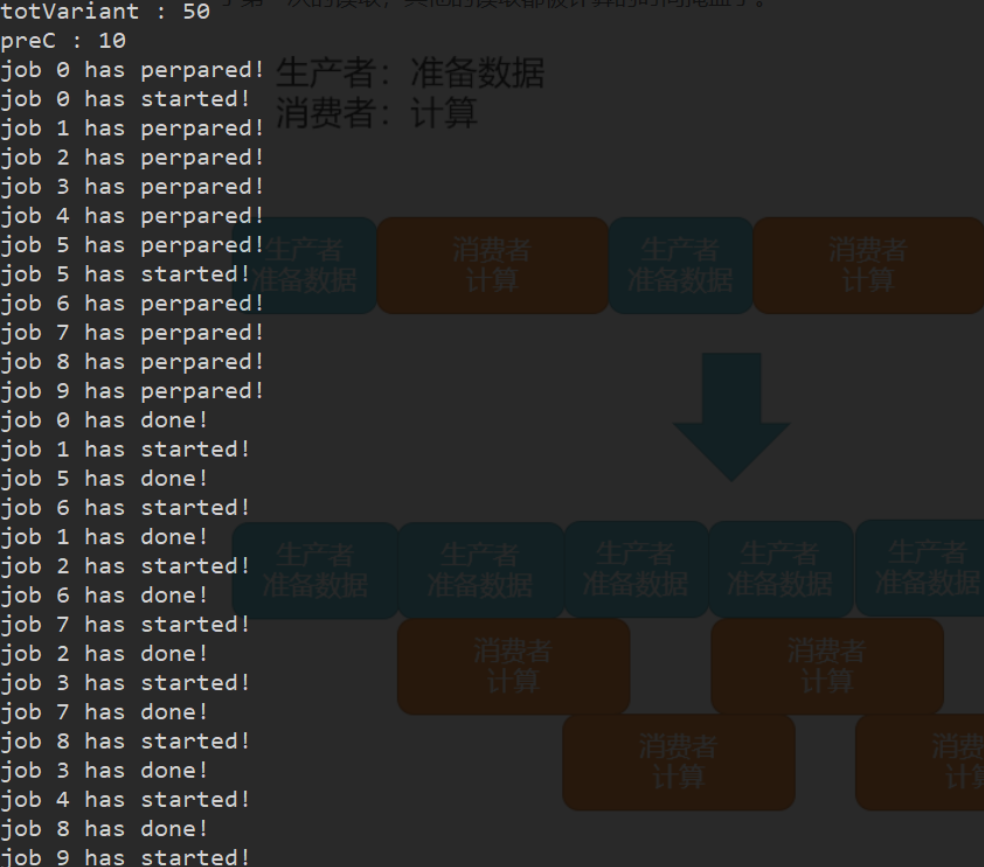
生产者消费者模型
根据上面的结果分析,发现随着核心数的增加,计算部分所占的时间越来越少,而准备数据的时间没有变,可能热点由计算部分转移到准备数据,这是我们不希望看到的。所以我们实现了下图所示的一种生产者消费者模型,线程0读完第一个数据,线程2就可以计算,此时线程1不停下,继续读下一个数据,读完传给线程2开始计算......这样除了第一次的读取,其他的读取都被计算的时间掩盖了。
代码实现:
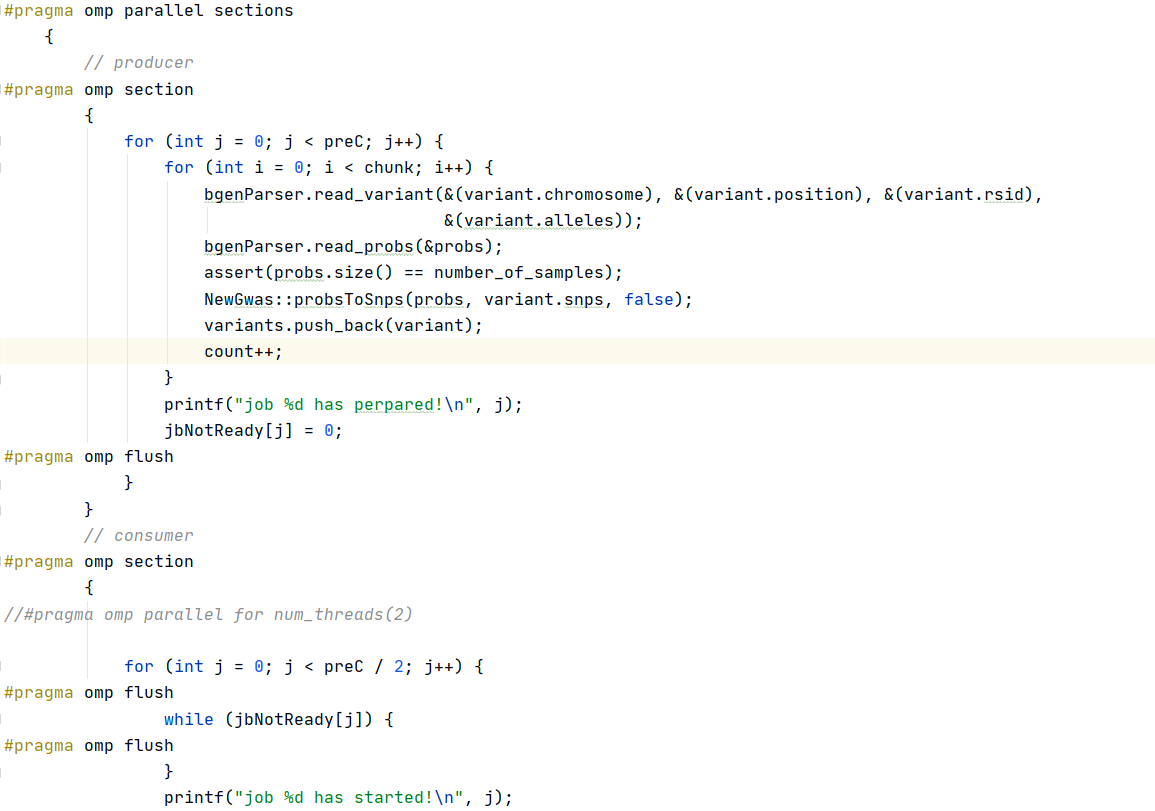

下面是运行过程的输出:
Update 2020.06.25
最近有毛概的考试,所以大部分时间看毛概了,不过对数据的解析也做了不少工作。
首先是对sample数据的解析,这部分相对来说稍微简单一点,就是对csv文件进行解析,首先看一下原始数据:

第一行是表头,表示一些表型,一共45种;第二行是表型的类型,具体的,分为 0,D,C,B,P,依次表示:id,Discrete covariate, Continuous covariate,Binary phenotype,Continuous phenotype
然后后面就是具体的数据了,其中NA表示未知数据
下面我们实现一个对sample数据的解析器:
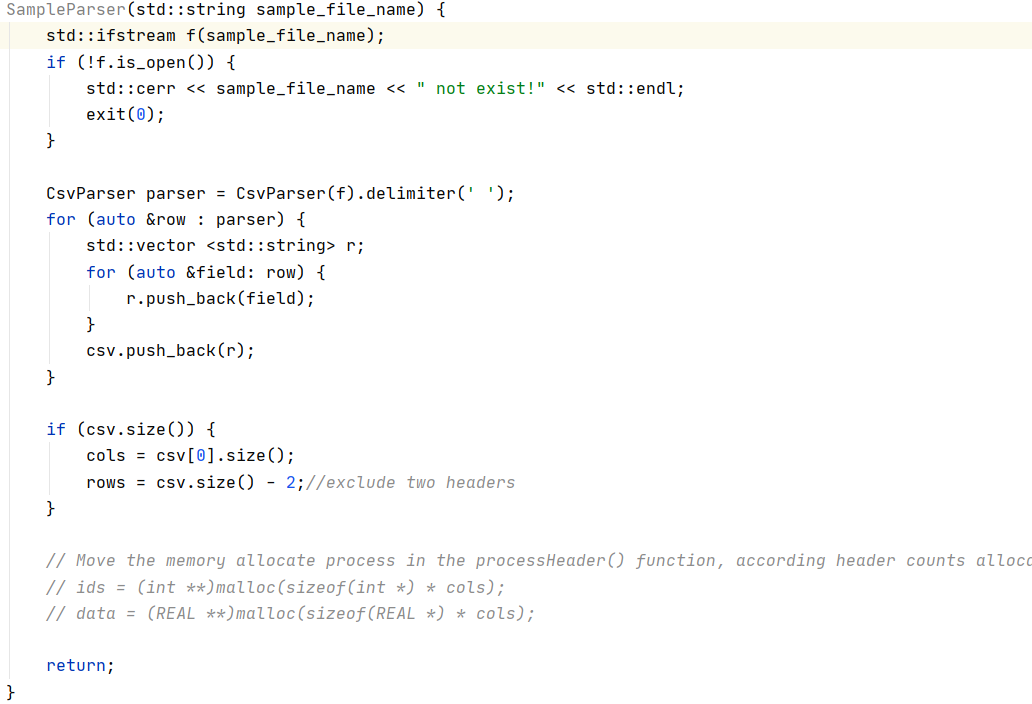
SampleParser就是我们实现的解析器class,上面展示的是他的构造函数部分,调用了csvparser.hpp中的函数,得到了cols和rows分别表示列数(表型数)和数据条数
其中rows=csv.size()-2这里减去的2就是首行表型name和第二行表型的type
然后动态的申请内存,不是静态的提前开好,而是用多少申请多少:

其中ids和data是指针的指针,后者是double类型的,然后根据行数动态的申请内存
下一步就是把初始的type转化成data该有的type,这里是double,这里输出一下处理的过程:

这些是表型name和type,然后和现在在看回归分析的同学沟通了一下,具体的分析方法是广义线性模型,输入要用到一个表型和其他几个协变量,这里在运行参数中设置了表型是sex,协变量是pca1:

以上基本上就是整个sample数据的解析过程了,这个是初始版本,其实有个可以多线程优化的解析csv文件的方式,这里没有用,虽然整个程序还没有写完但是预计热点大概率不在这里,多线程了也没有什么用处,暂时没有优化,后期如果发现这一步变成热点的话可以再修改。
这里用的是BGEN v1.2,它相比之前的版本做了一些改进,具体如下:
支持可变倍性和显式缺失数据。 支持多等位基因变体(例如复杂的结构变体)。 通过支持以可配置精度存储的基因型概率来控制文件大小。 支持存储样本标识符。
解析这种数据就必须找官方的文档参考了,因为初始文件是二进制,不像sample一样可以直接看到数据。
参考官方文档对其的解释:

一个BGEN文件由一个标题块和一个可选的样本标识符块组成,该标题块提供有关该文件的常规信息。这些之后是一系列连续存储在文件中的变体数据块,每个变体数据块均包含单个遗传变体的数据。
具体的:

即前4个字节存了一个偏移量,表示真正的数据(跳过标题块)从哪里开始(相对第5个字节的偏移量)。这样我们就能确定下标题块的大小以及数据块的位置。
然后根据标题块的详细描述对标题块先进性解析:

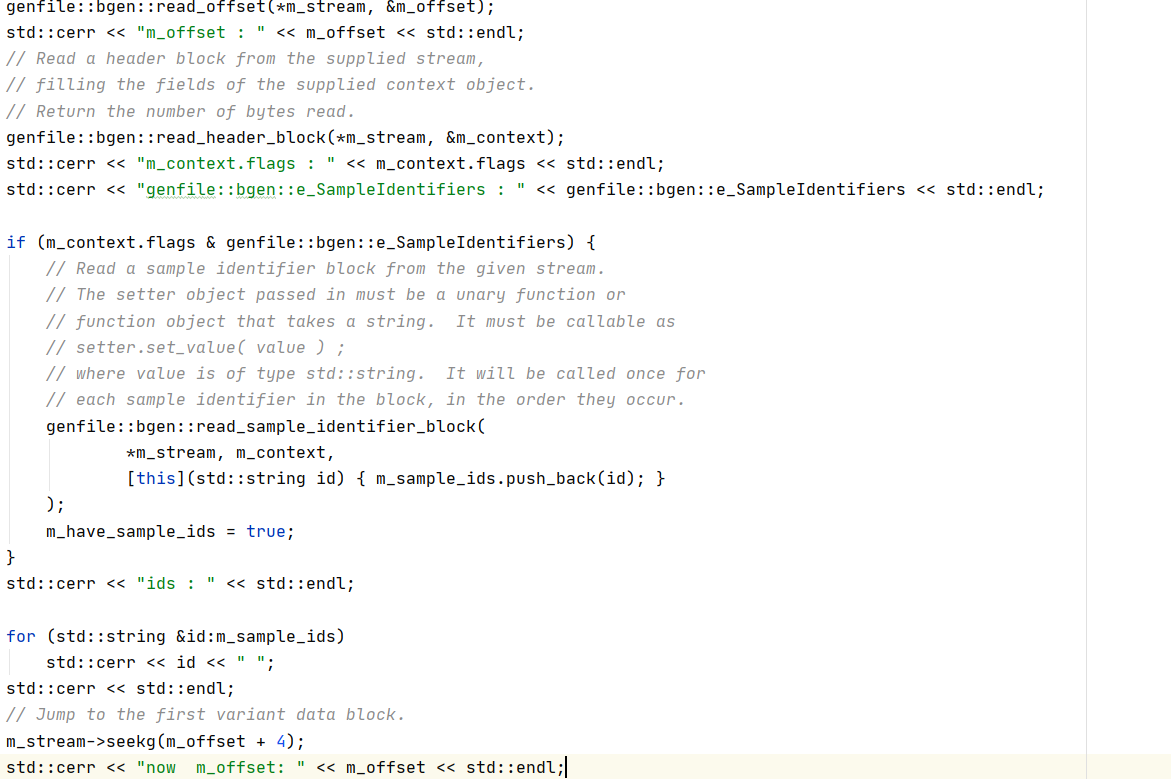
首先读出偏移量offset,然后读整个标题块,如果满足条件,就具体的读具体的样本表示块,最后把指针跳到具体的变异数据块。下面是这部分的输出:
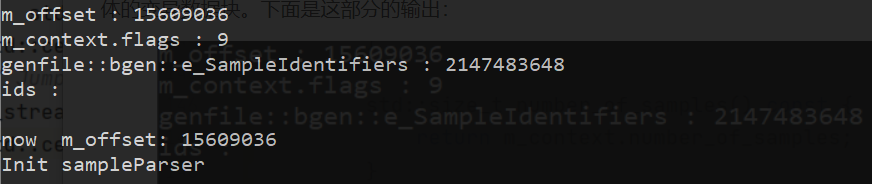
下面是具体的变异块的解析,还是按照官方的文档摸清楚他的文件结构
Update 6.25:
最终,基本理清楚每部分数据是干嘛的之后,把sample数据和bgen数据合并。
大体的思路是,首先50w个人,没人一行,然后每一行存这个人的相关信息,这里为了减少对内存的要求,并不是一次性把整个bgen文件读进来操作,而是切成一块一块的。
整个21号染色体的变异信息有37G,总共1261158条变异信息,调试部分用的数据是4个包含10条变异的bgen文件:

初步定下运行单个文件包含100条变异,后期在跑在更大的数据上:

经历数据解析之后的sample格式大体如下:

解析好的bgen格式(某一个变异):
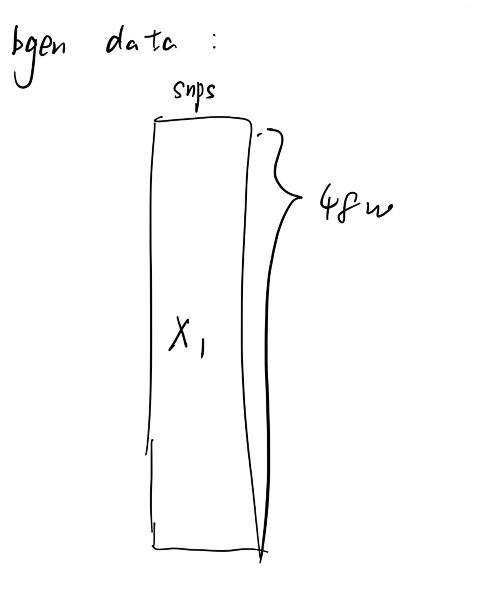
合并之后用于计算的数据格式:
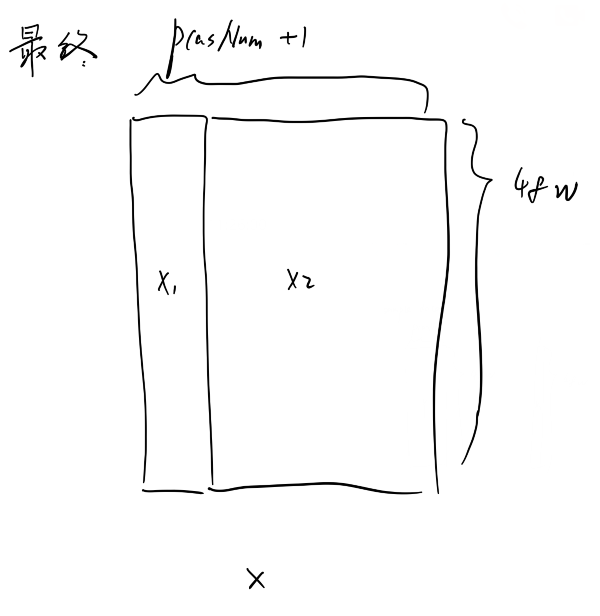
其中48w就是数据中的48w(接近50w)个人,pcasNum就是输入的协变量个数,+1是要分析的表型。
至此数据解析部分就全部完成了,不过没有加并行来优化,和之前的理由一样,整个程序的热点不一定在这里,而且很大的可能性不在这里,所以优化的重心放在后面计算部分。
核心代码:
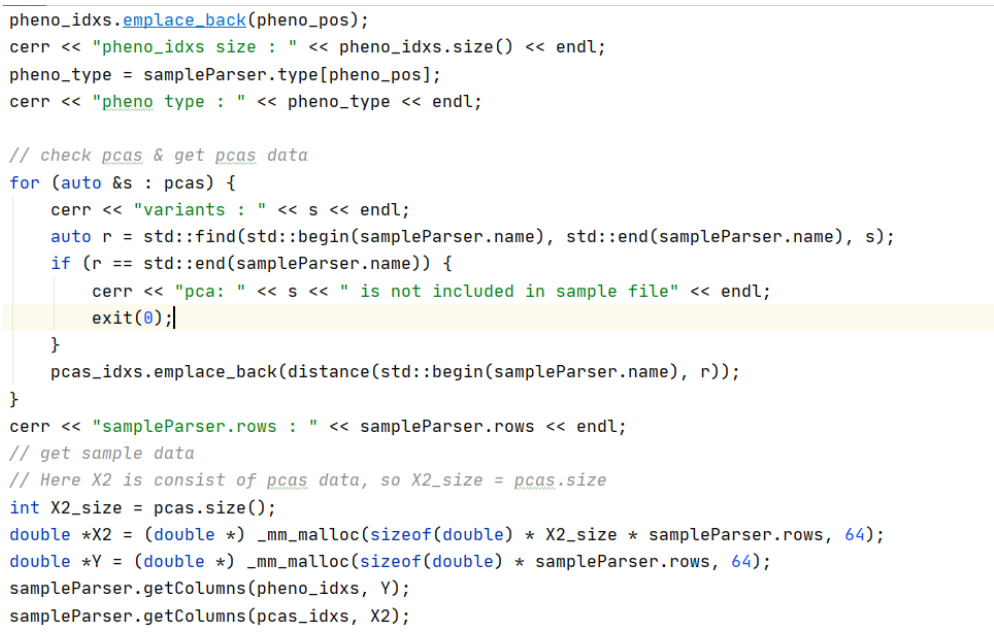

运行结果:

Update 2020.06.24



Update 6.16
由于染色体上的基因数据和变异太多了,存储是个大问题,所以采用压缩算法是十分有必要的,而且最好的无损的压缩算法。
现在下载好的21号染色体基因数据是经过zstd压缩的,要想解压缩,就要先了解这一种数据压缩算法。
- zstd是Facebook在2016年开源的新无损压缩算法,优点是压缩率和压缩/解压缩性能都很突出。
- 在我们测试的文本日志压缩场景中,压缩率比gzip提高一倍,压缩性能与lz4、snappy相当甚至更好,是gzip的10倍以上。
- zstd还有一个特别的功能,支持以训练方式生成字典文件,相比传统压缩方式能大大的提高小数据包的压缩率。
- 在过去的两年里,Linux内核、HTTP协议、以及一系列的大数据工具(包括Hadoop 3.0.0,HBase 2.0.0,Spark 2.3.0,Kafka 2.1.0)等都已经加入了对zstd的支持。
- 可以预见,zstd将是未来几年里会被广泛关注和应用的压缩算法。
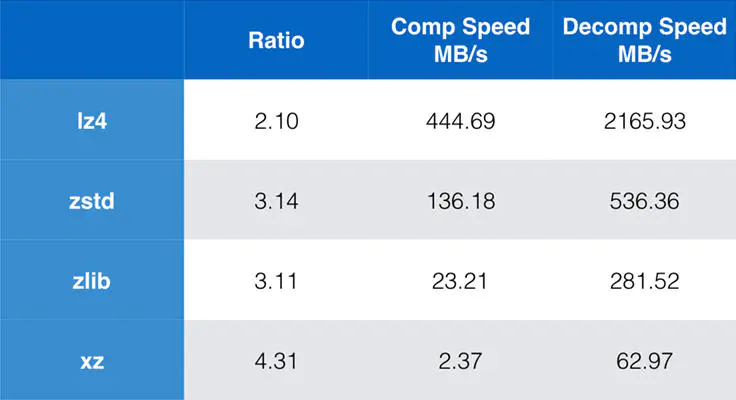
可以看到zstd虽然在压缩速度和压缩比上都有很好的表现,而且其特性恰好很适合基因数据的压缩。
关于项目的软件环境:
首先优化的话必不可少的是并行和高性能的数学库,并行暂时准备使用linux系统自带的pthreads和openmp,高性能数学库使用intel的mkl。
最近一直卡在intel编译器的配置上,直接一键安装了intel的parallel_studio_xe_2019,里面带有icc icpc mkl等,这里简单说一下配置过程:
首先去intel官网下载parallel_studio_xe_2019linux压缩包(会用到学生邮箱获取序列号),本地解压之后直接运行install.sh,按照提示一步步安装。
注意可能需要安装一些其他环境。
最终参照官方提供的文档配置环境:


这一步没有按照推荐的使用Eclipse,而是使用了Clion。
服务器上也是按照这个过程安装。
最终测试了一个vector的程序来比较g++和icpc在性能上的差异:

一个90s 一个2s,可以看到性能的提升还是十分明显的。
Update 2020.06.12
这几天下载数据花了不少的时间,因为数据太大了,21号染色体的基因信息有50G.
现在放到了服务器上,本地切下来一小部分用于初步的项目调试.最后在应用大数据进行关联分析.

其中dataxxx.bgen是基因数据,ukb_imp_chr_v3.sample是表形数据.后续的工作就是解析两种数据并进行关联分析.
具体的,sample是csv格式,首行存储列名,第二行存储数据类型.不同列分别存储了ID,表型,临床变量等信息.形式如图:
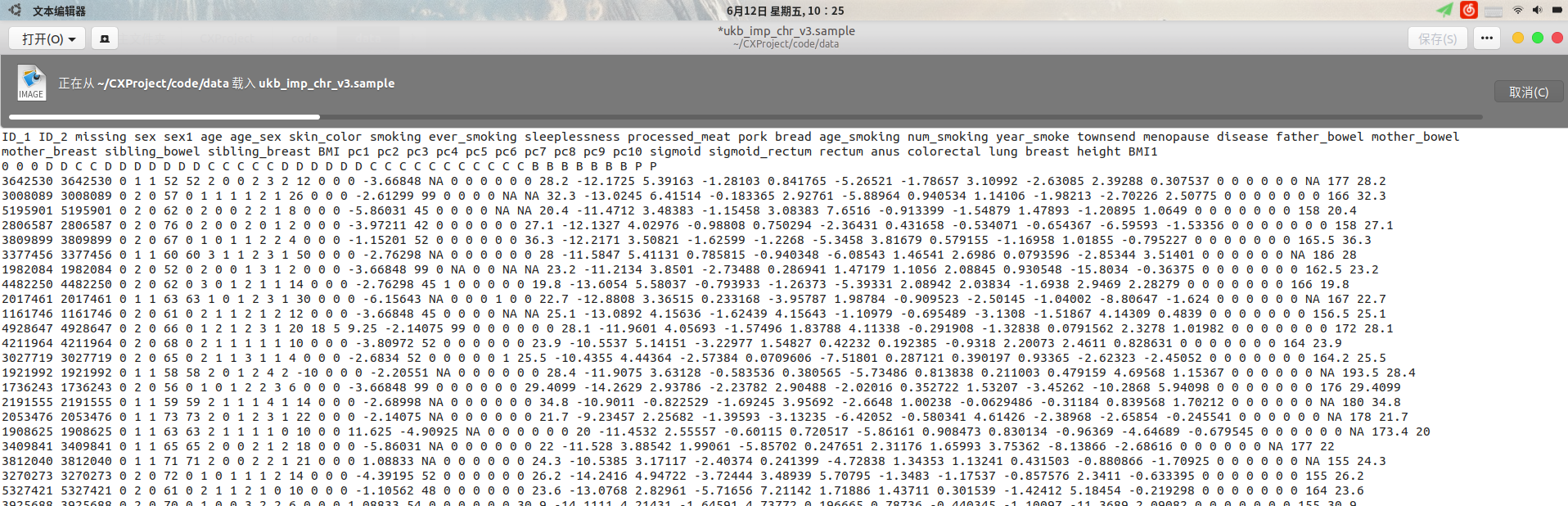
完整的21号染色体数据中共包含487409个个体的1261158个SNPs,这里的所有.bgen文件来自所有样本的21号染色体的变异信息,每个文件包含10个SNPs.